
Research Topics
New strategy for chemical reaction development using in silico screening

This work represents a strategy for reaction development using in silico reaction screening on a supercomputer with the artificial force induced reaction method, which led to the development of three-component cycloaddition reactions with difluorocarbene.
Background: Development Process of Chemical Reactions
Chemical reactions create molecules, which construct materials in our life such as products or pharmaceuticals. However, the development of chemical reactions has been primarily conducted through a trial-and-error approach that relies on the intuition and experience of each chemist. This conventional process requires an enormous amount of experiments, involving the corresponding energy and cost, and thus generates a lot of waste.
To solve this problem, great attention has been given to new strategies for the development of chemical reactions using computational and information sciences in recent years, and thus this research area has been studied around the world as the progress of CPU power and AI technology has been made. In this regard, if unknown chemical reactions could be predicted computationally from scratch, their development can be significantly accelerated. However, such a strategy has not been available because estimating all conceivable competitive reaction pathways is difficult using general computational methods.
Here we aimed to establish a new strategy for the development of chemical reactions, based on the computational reaction simulation with the artificial force induced reaction (AFIR) method, the core technology of the ERATO project, and a supercomputer. The AFIR method, a technique that can automatically search for reaction pathways using quantum chemical calculations, enables us to explore possible pathways including side reactions from the single input file. In combination with kinetic analysis, the calculated yield can be estimated as well. Hence, we started this project for demonstrating this proof of concept, which is the development of synthetically valuable reactions found via the AFIR-based screening by experiment.

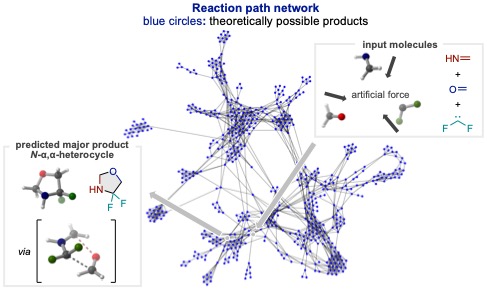
Approach: Reaction Screening by the AFIR Method
This study focused on the development of three-component reaction with difluorocarbene and a pair of unsaturated compounds (a pair from components bearing a C=O, C=C, C=N, or C≡C bond). First, we performed computational simulations of all combinations of ten reactions using the AFIR method to investigate what products would be obtained in each reaction. For practical calculations, we used the simplest molecules (i.e., formaldehyde, ethylene, methanimine, and acetylene) as the components bearing unsaturated bonds for calculations, which allowed us to explore the reaction pathways between each unsaturated bond and difluorocarbene while significantly reducing the calculation time.
The results of computational reaction screening suggested the cycloaddition reaction between "azomethine ylide"—a reaction intermediate generated from difluorocarbene and methanimine—and the third unsaturated bond component would proceed preferentially, furnishing a cyclic product bearing two fluorine atomsαto the nitrogen atom. Although compounds that bear a difluoromethylene group at the analogous β- or γ-positions are known as novel diabetes drugs with dipeptidyl peptidase 4 (DPP-4) inhibitory activity, analogous α-difluorinated compounds have rarely been studied and their synthetic methods are limited. Their efficient synthetic method would potentially contribute to the discovery of new biological activity. We, therefore, started the experiments to realize the computationally predicted reaction to generate α,α-difluorinated N-heterocycles.
Research Results: Realization of Three-Component Reaction
While simple unsaturated compounds were used for the calculations to reduce its cost, stable reagents and chemical species which can be generated in situ are needed in experimental investigation. Therefore, we performed the experiments with the available compounds which have been used as typical substrates in organic synthesis to realize the computationally predicted reactions. At this point, we also leveraged the AFIR method to screen the compounds to figure out the suitable substrates with low activation barriers for the predicted reactions, which led to the realization of the three-component dearomative cycloadditions of pyridines with benzaldehyde and difluorocarbene which was generated in situ, providing the desired fluorinated N-heterocycles in high yield.
Further experimental investigation revealed that the three-component reaction proceeded with a series of coupling partners such as ketones, imines, alkenes, or alkynes. As such, our group has successfully developed three-component reactions that provide diverse fluorinated N-heterocyclic motifs based on the computation, in which bioactive nicotine natural products could be used as pyridine substrates in the reaction.
In addition, competitive side reactions are also assumed in the reaction path network constructed by the AFIR method, which allowed us to predict the substrates which can undergo the present cycloaddition or not. For example, the reaction with benzaldehyde or acrylonitrile is expected to proceed because the activation barrier for the desired cyclization reaction is lower than that of the side reaction, and it was verified by the experiment. On the other hand, the reaction with ethylene or styrene, or the reaction with two molecules of benzaldehyde instead of a pyridine/electrophile, is not expected to proceed because the activation barrier of the side reaction is lower than that of each cycloaddition, and it was also verified. Thus, we have demonstrated that the AFIR method can predict the reaction performance.

Outlook: Computation-Driven Development of Chemical Reactions
This research describes that a series of procedures, in which experiments are conducted based on the reaction pathways predicted by the AFIR method, the core technology of ERATO project, was highly effective in developing a new reaction and its process was significantly accelerated. As such, we believe that this strategy will be used as a next-generation approach in various reaction development in the future.
Related Information
- Original Paper: Nat. Synth. 2022, DOI: 10.1038/s44160-022-00128-y
- Featured Pages: