理化学研究所(理研) 生命医科学研究センター 医科学数理研究チームの角田 達彦 チームリーダー(東京大学 大学院理学系研究科 生物科学専攻 医科学数理研究室 教授、東京医科歯科大学 難治疾患研究所 医科学数理分野 教授)らの国際共同研究グループは、人工知能技術の1つである「深層学習注1)」で扱えるように、ゲノミクス注2)データなどの非画像データを画像データに変換する方法を開発しました。
本研究成果により、遺伝子データなどさまざまな非画像データを深層学習で扱うことで、背後にある複雑な特徴や構造を抽出できるようになり、医療での診断や医学・生命科学など広範囲の応用に貢献すると期待できます。
ゲノミクスデータなどの多くのデータは非画像データであるため、深層学習の能力を生かすには画像データにする必要があります。
今回、国際共同研究グループは、遺伝子発現などのゲノミクスデータを使って医療診断や予測のためのクラス同定や分類を高精度に行うために、「適切な変数(画素)の配置」、「特徴抽出」、そして「適切な分類モデルの構築」という3つのステップを行う「DeepInsight(ディープインサイト)法」を開発しました。ディープインサイト法では最初の段階で、画素としての変数を適切に再配置し、非画像データから画像データへの変換を行うことで、非画像データの解析に対しても深層学習が可能になりました。この新手法を、がん遺伝子発現などの実データに適用した結果、既存のランダムフォレスト法注3)などの機械学習よりもはるかに高精度で分類できることが分かりました。本方法論には普遍性があり、さまざまな非画像データを深層学習で扱うことができます。
本研究は、英国のオンライン科学雑誌「Scientific Reports」(2019年8月6日付け:日本時間8月6日)に掲載されます。
本研究は、科学技術振興機構(JST) 戦略的創造研究推進事業(CREST)「科学的発見・社会的課題解決に向けた各分野のビッグデータ利活用推進のための次世代アプリケーション技術の創出・高度化(研究総括:北海道大学 田中 譲)」における研究課題「医学・医療における臨床・全ゲノム・オミックスのビッグデータの解析に基づく疾患の原因探索・亜病態分類とリスク予測(研究代表者:角田 達彦)」、文部科学省 新学術領域研究「代謝アダプテーションのトランスオミクス解析(領域代表者:東京大学 黒田 真也)」の「次世代ヒト全ゲノム・オミクスの解析方法論の開発と応用(研究代表者:東京大学 角田 達彦)」、東京医科歯科大学 難治疾患研究所 国際共同研究拠点活動による支援を受けて行われました。
<背景>
ゲノムなどのゲノミクスデータは、病気などの個人差の解析・診断に役立つと考えられています。しかしゲノミクスデータは、数万から数千万の変数を持つ超高次元データであることが多く、伝統的な統計学では解析が難しい課題が多く存在します。これを克服するために、近年では「機械学習」に期待が集まっています。
機械学習の1つの応用例として、機械が多くの変数の中から重要と判断した変数群を抽出し、その変数群を用いて、新規のサンプルが特定の病気と関連があるか否かを判別することが挙げられます。ところが、ゲノミクスデータには背後に複雑な構造があることから、典型的な機械学習では簡単に取り扱えません。機械学習の多くでは、そのような複雑な構造はあらかじめ解いて、互いに独立な因子にした(特徴抽出)後の段階から使うと、精度が高くなる場合が多いです。ということは、前段階の特徴抽出をいかにうまく行うかがポイントになります。
そこで国際共同研究グループは、その特徴抽出を自動的に行えないかと考え、機械学習の中でも、現在の人工知能技術の主流の1つであり、特徴抽出能力を自前で持つ「深層学習」に着目しました。
深層学習を行うモデルの1つである「畳み込みニューラルネットワーク(CNN)注4)」では、サンプルは画像の形で入力し、隠れ層によって特徴抽出と分類を行います。このモデルではデータから自動的に特徴を導き出すため、追加の特徴抽出手法は必要ありません。また、画像に含まれる複雑な構造が見つけられることから、自動運転などへの産業応用も見込まれています。
実際の空間では多くの場合、近接する部分同士には何らかの類似性があります。同様に、写真や絵などの画像にも、互いに近接する画素は類似の情報を持つ傾向にある、という秩序があります。CNNでは、画素ごとに処理を行う際に周囲の情報も使って処理するため、画像内の構造を捉え、特徴を抽出することが可能になります。もし画素の配置が不適切だと、CNNの特徴抽出とその後の分類性能に悪影響が出る可能性があります。
CNNによりゲノミクスデータを解析するためには課題があります。ゲノミクスデータなどの多くのデータは非画像形式であり、隣り合う変数同士では明確な関係性が見られない場合が多くあります。CNNは入力として画像データを必要とするため、ゲノミクスデータなどを直接は使用できません。ただし、データ同士の関連を適切に考慮しつつ非画像データを画像データに変換できれば、CNNを使って特徴抽出と学習ができ、典型的な機械学習よりも分類性能を向上させることが期待されます。そのためには、画素としての変数の配置をうまく行う方法を考える必要があります。
<研究手法と成果>
国際共同研究グループは、データを使って診断や予測のためのクラス同定・分類を高精度に行うためには、「適切な変数(画素)の配置」、「特徴抽出」、そして「適切な分類モデルの構築」、という3つのステップを経る必要があると考え、それらを全て統合した「DeepInsight(ディープインサイト)法」を提案しました。
まず、変数の配置に関して、近傍情報を無視して独立に変数を扱うよりも、類似した変数や生データをクラスターとして変換する方が、周囲の重要な情報を互いに補完・統合できるため、信頼性が高くなります。そこで、ディープインサイト法では最初に、似た変数をまとめて配置し、異なるものを離して配置することによって、隣接する変数をまとめて画素集合のように使えるようにした画像を作成します。
例として、図1aに遺伝子発現値からなる変数ベクトルxを変換Tで変数行列Mに変換する全体像を示します。変数ベクトルの変数の位置は、変数の類似性によって決めます。例えば、計算の結果類似性が高い変数g1、g3、g6、gd同士が互いに近くになるように、各変数の位置をマトリックス内で決めます。そしてそれらの変数の値を各位置に代入します。これにより、サンプルごとに1つずつ画像が生成されます。つまり、d次元変数を持つN個のサンプルが、m×n行列を持つN個のサンプルに変換されます。
図1bに、より具体的な手順を示します。まず、学習セットにt-SNE注5)やカーネル主成分分析(kPCA)注6)などの類似度計算手法や次元圧縮手法を用いて、2次元平面を得ることで、各変数の位置(点)を決めます。CNNに入力するために、全ての点を含む最小の長方形を見つけ、回転させて水平・垂直の形式にし、各座標をピクセルにマッピングします。変数ベクトルを画像に変換すると、CNNで特徴抽出をし、分類や予測することができるようになります。
図2aに、ディープインサイト法により、2種類のがん検体を変換した画像を示します。今回、CNNのアーキテクチャー(論理的構造)として、効率的にモデルを学習させるため、異なるフィルタサイズを同時に扱えるものを設計しました(図2b)。
図3に実際のデータを変換した例として、異なる種類のがん検体の遺伝子発現データ、異なる種類のテキストデータ、異なる種類の母音データを画像に変換したものを示します。このようにして学習を終えると、新しいサンプルに対してもクラスを決定できます。
次に、ディープインサイト法の検証を試みました。そのため、「遺伝子発現データ」、「テキストデータ」、「母音データ」、「2つの人工データ」のセットを用いて、既存の最先端の分類手法であるランダムフォレスト法、決定木注7)、アダブースト法注8)などと結果を比較しました。特に遺伝子発現データは、TCGA注9)(https://www.cancer.gov/ccg/research/genome-sequencing/tcga)からの公共データセットであり、10種類のがんに対応するRNA-seq注10)遺伝子発現データの6,216のサンプルで、各サンプルには60,483の遺伝子発現値(変数)が付けられています。
その結果、RNA-seqデータのテストセットを用いたとき、ランダムフォレスト法では分類精度96%だったのに対し、ディープインサイト法では99%を達成しました。母音データでは、ランダムフォレスト法の分類精度90%に対してディープインサイト法は97%、テキストデータでは、ランダムフォレスト法の分類精度90%に対し、ディープインサイト法は92%の精度を達成しました。残りの2つの人工データセットでも同様の結果でした。
さらに、5つのデータセット全てを用いて平均分類精度を計算した結果、既存の技術のうち最高だったランダムフォレスト法の86%に対し、ディープインサイト法は95%の平均分類精度を記録し、はるかに優れたものであることが分かりました。
<今後の期待>
本手法により、非画像データの解析に対しても、深層学習、特にCNNの能力を活用することができます。すなわち、特徴抽出、次元削減、疎なデータや超高次元データからの隠れた構造の発見、少ないデータで学習するためのデータの水増しとアップサンプリング、ラベル付き/ラベルなしサンプルによる半教師あり学習注11)、そして時系列データ解析などです。また、DNA配列やたんぱく質配列、RNA-Seqなどのさまざまなオミクスデータを深層学習によって解析できる可能性があります。
さらに、現時点で本手法は、入力が単層で2次元行列型のCNNを想定していますが、これを複数の層を組み込むように拡張すれば、例えば、遺伝子発現、メチル化、突然変異などのマルチオミクスデータを扱う問題にも適用することができます。深層学習がより利活用されることにより、医学・生命科学の複雑なデータがひもとかれ、将来の個人ごとの診断や予測に役立てられることが期待できます。
<参考図>
-
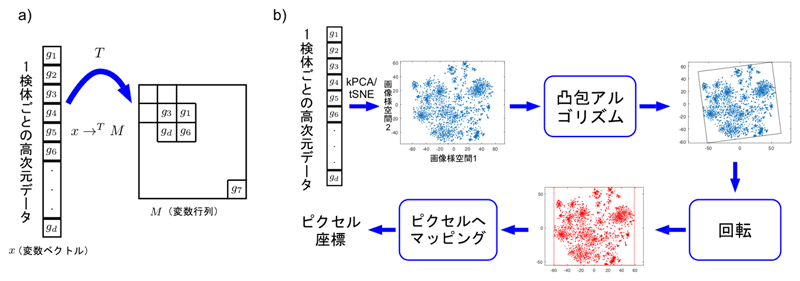
図1 変数ベクトルxを変換Tで行列に変換する全体像と変換の具体的な手順 - a)遺伝子発現値からなる変数ベクトルxを、変換Tで変数行列Mに変換する。類似性をもとに判断した結果、変数g1、g3、g6、gdが互いに近くになるように各変数の位置を右側のマトリックス内で決める。そしてそれらの変数の値を各位置に代入すると、検体ごとに1つずつ画像が生成される。つまり、d次元変数を持つN個のサンプルが、m×n行列を持つN個のサンプルに変換される。
- b)変換の具体的手順として、学習セットにt-SNEやカーネル主成分分析(kPCA)などを用いて、2次元平面を得ることで、各変数の位置(点)を決める。CNNに入力するために、凸包アルゴリズムで全ての点を含む最小の長方形を見つけ、回転させて水平・垂直の形式にし、各座標をピクセルにマッピングする。
-
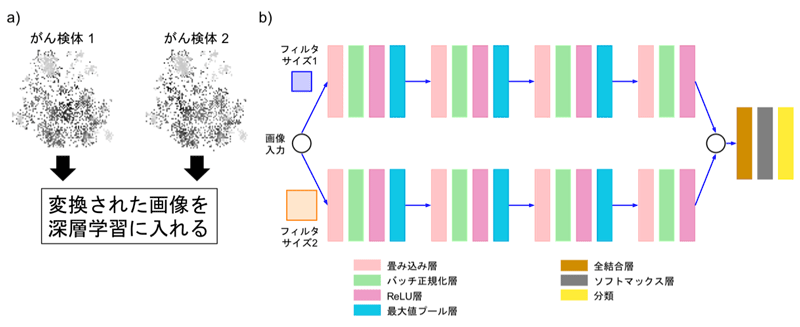
図2 ディープインサイト法で変換したサンプルの像と使用した深層学習のアーキテクチャー - a)2種類のがん検体を、図1で示したディープインサイト法で変換した画像を深層学習に入れる。
- b)設計したCNNアーキテクチャー。画像を入力データとし、分類結果を出力するように学習する。効率的にモデルを学習させるため、異なるフィルタサイズを同時に扱えるように上下2段の構成にした。
-
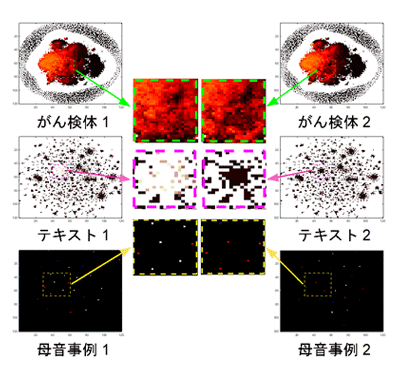
図3 図1の方法で変換したサンプルの画像の実例 上段はがん、中段はテキスト、下段は母音のそれぞれの非画像データを画像データに変換した。
<用語解説>
- 注1)深層学習
- 多層のニューラルネットワーク(ディープニューラルネットワーク)による機械学習手法。ディープラーニング。
- 注2)ゲノミクス
- ゲノムや遺伝子について研究する生命科学分野。
- 注3)ランダムフォレスト法
- ランダムサンプリングしたトレーニングデータと説明変数を用いて、数千~数万の決定木を作り、各決定木の予測結果の多数決もしくは平均を取ることで、最終結果を決定する集団学習アルゴリズム。
- 注4)畳み込みニューラルネットワーク(CNN)
- 特に画像の分類や識別で高い性能を発揮するディープラーニングの1つ。あらかじめ与えられていた画像データから画像の特徴量を直接抽出し、ネットワークを学習する。CNNはConvolutional Neural Networkの略。
- 注5)t-SNE
- t分布型確率的近傍埋め込み法(T-distributed Stochastic Neighbor Embedding)。高次元データを、2次元などの低次元空間に変換する、非線形の次元削減手法。類似したデータ点同士が確率的に近くになるように対応付ける。
- 注6)カーネル主成分分析(kPCA)
- データをカーネル法によって非線形変換した後に主成分分析を行う方法。非線形なデータの場合には、主成分分析よりも適切な次元削減が行われる可能性が高い。kPCAはKernel Principal Component Analysisの略。
- 注7)決定木
- 木構造を用いて分類や回帰を行う機械学習手法の1つ。もともとは決定を行うための分岐のグラフを指すが、観察データから目標値を導く予測モデルや手法を指すことも多い。
- 注8)アダブースト法
- 弱い分類器を多数用意し、データ全体が最も説明できるように各分類器の重みを調整することにより、それらの組み合わせである分類器全体が強くなるように学習する方法。
- 注9)TCGA
- The Cancer Genome Atlas(がんゲノムアトラス)。米国の大規模がんゲノムプロジェクトで、20種類以上のがん種に対して、ゲノム、エピゲノム、遺伝子発現、たんぱく質の変化などが網羅的に調べられ、データが公開もしくは制限共有されている。
- 注10)RNA-seq
- いわゆる「次世代シークエンサー」による実験によって取得された、RNAの配列断片データ。これをもとに、遺伝子などの転写産物の発現量や配列、スプライスバリアントなどが解析できる。
- 注11)半教師あり学習
- 学習のために外から正解を与える教師ありデータと、外から正解を与えない教師なしデータを、混在させて効率的に学習する方法。教師ありデータを収集するにはコストがかかり過ぎるため、少ない教師ありデータと、多くの教師なしデータが得られる状況がある。その場合、多くの教師なしデータが、データの分布などの情報を補うことによって、分類や回帰の精度が良くなることがある。
<論文タイトル>
- “DeepInsight: A methodology to transform a non-image data to an image for convolution neural network architecture”
- DOI:10.1038/s41598-019-47765-6
<お問い合わせ先>
-
<研究に関すること>
角田 達彦(ツノダ タツヒコ)
理化学研究所 生命医科学研究センター 医科学数理研究チーム チームリーダー
東京大学大学院 理学系研究科 生物科学専攻 医科学数理研究室 教授
東京医科歯科大学 難治疾患研究所 医科学数理分野 教授
Tel:045-503-9556 Fax:045-503-9555
E-mail:tatsuhiko.tsunoda riken.jp
riken.jp -
<JST事業に関すること>
舘澤 博子(タテサワ ヒロコ)
科学技術振興機構 戦略研究推進部 ICTグループ
Tel:03-3512-3524 Fax:03-3222-2064
E-mail:crestjst.go.jp -
<報道担当>
理化学研究所 広報室 報道担当
Tel:048-467-9272 Fax:048-462-4715
E-mail:ex-pressriken.jp東京大学 大学院理学系研究科・理学部 広報室
Tel:03-5841-0654 Fax:03-5841-1035
E-mail:kouhou.sgs.mail.u-tokyo.ac.jp東京医科歯科大学 総務部 総務秘書課 広報係
Tel:03-5803-5833 Fax:03-5803-0272
E-mail:kouhou.admtmd.ac.jp科学技術振興機構 広報課
Tel:03-5214-8404 Fax:03-5214-8432
E-mail:jstkohojst.go.jp