ポイント
- 強力ながん化能を有するがん遺伝子(横綱がん遺伝子)を抑制する薬剤は優れたがんの分子標的治療薬となるが、そのような遺伝子はわずかしか同定されていない。
- RACファミリー遺伝子が、突然変異により横綱がん遺伝子になることを発見。
- 乳がんや悪性黒色腫でRAC遺伝子変異が存在。新しいがんの分子標的治療薬へ。
JST 課題達成型基礎研究の一環として、自治医科大学の間野 博行 教授(兼 東京大学 特任教授)らは、乳がんや悪性黒色腫の原因となる強力ながん化能を有するがん遺伝子(横綱がん遺伝子注1))を発見しました。
間野教授らは、2007年に肺がんの原因遺伝子EML4-ALK注2)を発見し、そのALK阻害剤は著明な治療効果をもたらし、日本でも2012年に治療薬として承認され臨床適用されています。このことは、横綱がん遺伝子を発見すれば、有効な分子標的治療薬が速やかに開発されることを意味しています。
今回研究グループは、発がん能力を持つ遺伝子を同定する機能評価法と、高精度次世代DNAシーケンサー注3)を用いた遺伝子変異スクリーニング法をそれぞれ開発しました。そして、それらを組み合わせることで、ヒトのがん細胞の中で「発がん機能」と「配列異常」を持つ遺伝子があるかを解析しました。その結果、ヒト線維肉腫細胞株から低分子量Gたんぱく質注4)の1つであるRAC1を発現するRAC1遺伝子の突然変異を発見しました。具体的には、RAC1たんぱく質の92番目のアミノ酸であるアスパラギンがイソロイシンに置換される変異です。さらに、このRAC1遺伝子の抑制により、ヒト線維肉腫細胞が細胞死を起こしたことから、RAC1遺伝子に変異が起こることがヒト線維肉腫細胞株の本質的な発がん原因であることが分かりました。また、ほかのヒトがんにおいてもRAC遺伝子ファミリーの変異があるか調べたところ、乳がんや悪性黒色腫など5種類のがんで変異があることも明らかにしました。
これら変異RACたんぱく質の機能を抑制する薬剤は、変異があるがん患者に対して有効な、全く新しいがん分子標的療法をもたらすと考えられ、その診断法とともに速やかな開発が期待されます。
本研究成果は、米国科学雑誌「米国科学アカデミー紀要(PNAS)」のオンライン速報版で2013年2月4日の週(米国東部時間)に公開されます。
本成果は、以下の事業・研究領域・研究課題によって得られました。
戦略的創造研究推進事業 研究加速課題
| 研究課題名 |
「新規がん遺伝子同定プロジェクト」 |
| 研究代表者 |
間野 博行
(自治医科大学 分子病態治療研究センター 教授/東京大学 大学院医学系研究科 特任教授)
|
| 研究期間 |
平成21年1月~平成26年3月 |
上記研究課題では、研究代表者らが開発した遺伝子機能スクリーニング法と次世代DNAシーケンサーを用いたゲノミクス解析法とを組み合わせることで、がん臨床検体から発がん原因遺伝子を同定し、その知見に基づく新たな分子診断法・分子標的療法を開発することを目指しています。
<研究の背景と経緯>
がんは現在先進諸国における成人の最大の死因であり、日本においても3人に1人ががんでなくなる時代となっています(厚生労働省「人口動態統計」)。旧来の抗がん剤による治療効果は限界に近づきつつあり、細胞の増殖関連たんぱく質を標的とした分子標的治療に期待が集まっています。しかし、残念ながら単剤治療で患者予後を改善できる分子標的治療薬はごくわずかしか存在しません。
本研究グループは2007年に、肺がんの一部で2番染色体内の微小な転座が生じた結果、EML4-ALK融合がん遺伝子が生じることを発見しました(図1、参考論文1)。正常ALKたんぱく質は細胞の増殖を司る酵素ですが、融合がん遺伝子から産生されるのは、その酵素領域が微小管会合たんぱく質EML4と融合した異常な酵素です。EML4-ALK融合がん遺伝子は極めて強力ながん化能を持っており、がんを誘導する遺伝子「がん遺伝子」の中でも特に強い「横綱がん遺伝子」というべきものです。ALK酵素活性を選択的に抑えるALK阻害薬は、EML4-ALK融合がん遺伝子陽性肺がんの全く新しい分子標的治療薬になると期待され、実際単剤治療による目覚ましい臨床効果が報告されています(参考論文2)。また、最初に臨床試験に入ったALK阻害剤クリゾチニブは2011年に米国で、また2012年には日本でそれぞれ承認販売されました。2007年のEML4-ALK融合がん遺伝子の発見からわずか4年後の薬剤認可は、世界のがん治療薬開発史上最速といえます。
この事実は、各種がんにおいて発がんに本質的な「横綱がん遺伝子」を発見すれば、その機能を抑制する薬剤が新しい特効薬となることを示唆しています。しかし、横綱がん遺伝子を効率よく発見するためには新しいスクリーニング技術の開発が必須でした。
本研究グループは、EML4-ALK融合がん遺伝子発見に至った独自の機能評価法に加えて、今回新たに次世代DNAシーケンサーを用いた高精度遺伝子変異スクリーニング法を開発し、両者を統合してがん細胞を解析することを試みました。その結果、ヒト線維肉腫細胞株から新しい横綱がん遺伝子「変異RAC1」を発見することに成功しました。
<研究の内容>
本研究グループは、まず次世代DNAシーケンサー解析のエラー率を下げる手法を開発し、各種がん検体・細胞株に対し網羅的配列解析法と、研究グループ独自の機能評価法の両者を行うことで「配列異常」があり、かつ「発がん機能」がある遺伝子を効率よく同定することを試みました。その結果、ヒト線維肉腫細胞株HT1080から、2種類の異常遺伝子を発見しました。1つはNRASたんぱく質の61番目のアミノ酸であるグルタミンがリジンに置換したNRAS(Q61K)、もう1つはRAC1たんぱく質の92番目のアミノ酸であるアスパラギンがイソロイシンに置換したRAC1(N92I)です(図2A)。また、両遺伝子変異によるがん化を確かめたところ、どちらにも認められました(図2B)。
NRAS、RAC1両遺伝子の産物であるたんぱく質は、どちらも低分子量Gたんぱく質であり、GDP注5)が結合した不活化状態とGTP注6)が結合した活性化状態の間を移り変わる、「細胞内スイッチ」の役割を果たしています。両遺伝子のうちNRAS遺伝子は、HRAS遺伝子、KRAS遺伝子とともにRASファミリー注7)と呼ばれる仲間の1つであり、いずれもさまざまながんにおいてゲノム点突然変異の結果、アミノ酸置換を生じて活性化されることが知られています。実際NRAS(Q61K)は、NRAS遺伝子のがん化変異として最も高頻度に生じるタイプです。ところが、RASファミリー以外の低分子量Gたんぱく質のがん化変異は知られていませんでした。今回、RAC1(N92I)ががん化能を持つことが明らかになりましたが、これは世界で初めてです。
同じ低分子量Gたんぱく質を産生するNRAS遺伝子とRAC1遺伝子のがん化変異が同一細胞に存在することは驚きでしたが、さらにどちらのがん遺伝子がヒト線維肉腫細胞株HT1080に必須の横綱がん遺伝子なのかを、遺伝子特異的なsmall interfering RNA(siRNA)注8)を用いて、HT1080細胞内におけるおのおのの遺伝子発現を選択的に抑制する実験で検証しました。その結果、RAC1遺伝子を抑制したところ速やかな細胞死が誘導されました(図3A)。ところが、NRAS遺伝子を抑制しても細胞増殖への影響は軽微であり、しかもRAC1遺伝子とNRAS遺伝子の両者を同時に抑制しても、RAC1遺伝子単独抑制時と変わりありませんでした。つまり、NRAS(Q61K)ではなくてRAC1(N92I)こそが、HT1080細胞における本質的な発がん原因であると考えられます。さらに、HT1080細胞にRAC1遺伝子のsiRNAを導入した後、変異のない野生型RAC1遺伝子を導入しても細胞増殖は回復しませんが、RAC1(N92I)を導入すると細胞増殖は回復しました(図3B)。この結果は、RAC1遺伝子のsiRNAを用いた細胞増殖の抑制効果が、間違いなくRAC1(N92I)の減少によるものであることを証明しています。また、RAC1たんぱく質の活性阻害薬をHT1080細胞培養液に添加しても、その濃度依存性に細胞死が誘導されたので、RAC1(N92I)が適切な治療標的であることが確認されました(図3C)。
RAC1遺伝子はRACファミリーと呼ばれるグループの一種であり、ほかにRAC2、RAC3遺伝子などを含みます。そこで、RAC1、RAC2、RAC3遺伝子の変異の有無について、さまざまながんの細胞株を次世代DNAシーケンサーで解析するとともに、既存のがん変異データベースなども確認したところ、RAC1遺伝子上に4種類、RAC2遺伝子上に5種類のアミノ酸置換を伴う変異を確認しましたが、そのうちがん化能が存在したのは表1に示す計5種類でした。特に、悪性黒色腫の約5%に変異RAC1遺伝子が存在するのは重要です。また、乳がんの中でエストロゲン受容体陰性、プロゲステロン受容体陰性、HER2陰性の予後不良群「トリプルネガティブ乳がん注9)」の一部でRAC遺伝子変異が発見されたことは、これら変異陽性の乳がん症例にとって重要な知見といえます。臨床検体を併せて解析した結果、RAC1/RAC2遺伝子のトリプルネガティブ乳がんにおける変異率は約3%でした。さらに、慢性骨髄性白血病の急性転化期から樹立した細胞株でも、変異RAC2遺伝子が発見されました。慢性骨髄性白血病の治療薬であるイマチニブが有効である慢性期から、どのような分子メカニズムによってイマチニブが無効な急性転化期へ移行するかは、ほとんど分かっていません。今回の活性型RAC2たんぱく質変異の発見は、慢性骨髄性白血病の急性転化に新しい知見をもたらしただけでなく、同様な症例に対する「イマチニブ+RAC阻害剤」という新しい治療法の可能性を示唆するものといえます。
一般に、低分子量Gたんぱく質はGDPが結合している不活性型(スイッチのオフ)と、GDPが離れて新たにGTPが結合した活性型(スイッチのオン)の間を移り変わっています(図4A)。がん化RAS変異たんぱく質の場合は全て、一旦結合したGTPが離れなくなる変異体であることが知られており、そのために恒常的にスイッチがオンされていると言われています(図4B)。一方、今回のRAC変異たんぱく質について解析したところ、全く異なったメカニズムで活性化されていました。通常、GDP結合型からGTP結合型へ移行する際には一連の専用酵素によってその反応が触媒されますが、今回発見したRAC変異体たんぱく質は全て、専用酵素がなくても自然にスイッチのオン状態へ移行する特徴を持っていました(図4C)。一方、一旦結合したGTPが離れる速度は野生型RACと変わりありませんでした。臨床検体において、この様なメカニズムで低分子量Gたんぱく質が活性化されることは今回初めて明らかになりました。
<今後の展開>
新たながん遺伝子として、RACファミリー変異体がさまざまながんで発見されました。HT1080細胞株における解析結果から、これら変異体は発がんの本質的な「横綱がん遺伝子」と考えられますので、その機能あるいは下流分子の活性を抑制する薬剤は、これら変異陽性がんの全く新しい、しかも有効な分子標的治療薬になると期待されます。
またがん化RAS遺伝子変異が、広くヒトのがんで存在しているにもかかわらずRAS阻害物質はこれまでの臨床試験で有効性が示されず、その理由が不明なままでした。今回のHT1080細胞株の解析結果は、変異RAS遺伝子は横綱がん遺伝子ではないので、ほかの横綱がん遺伝子と協同で発がん原因となっていることを示しています。そのようながんでは、RASたんぱく質の機能を抑制するだけでは十分な治療効果が得られませんので、RAS変異がんの治療薬開発のためには、共存するほかのがん遺伝子を明らかにすることが重要であることも分かりました。
<参考図>
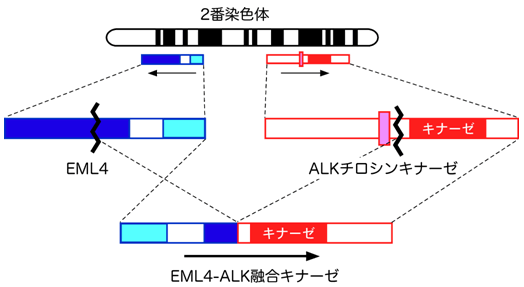
図1 EML4-ALK融合遺伝子
EML4遺伝子とALK遺伝子は、正常細胞においてどちらもヒト2番染色体上のごく近い位置に互いに反対向きに存在するが、両遺伝子を挟む領域が逆位となることでEML4-ALK融合遺伝子が生じる。その結果微小管会合たんぱくEML4のアミノ末端側約半分と受容体型チロシンキナーゼALKの細胞内キナーゼ領域とが融合し、極めて強いがん化能を持つ活性型EML4-ALKキナーゼが肺がんの中で産生されることになる。
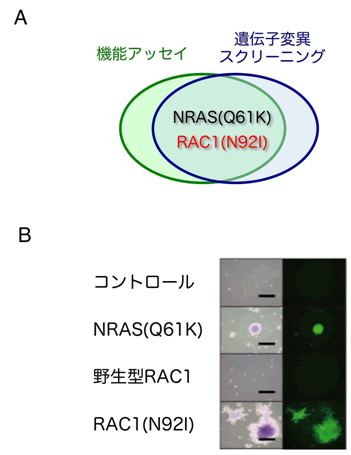
図2 RAC1がん遺伝子の発見
- (A)ヒト線維肉腫細胞株HT1080に対してレトロウィルスを用いた機能評価(機能アッセイ)と、次世代DNAシーケンサーを用いた遺伝子変異スクリーニングの両者を行い、どちらも陽性な遺伝子としてNRAS(Q61K)変異とRAC1(N92I)変異を発見した。
- (B)正常マウス3T3線維芽細胞株(コントロール)あるいは野生型RAC1導入3T3を寒天中で培養しても足場非依存性増殖は認められないが、NRAS(Q61K)導入細胞およびRAC1(N92I)導入細胞はいずれも明瞭に足場非依存性増殖が確認される(左パネル)。また遺伝子発現ウィルスが同時にGFPたんぱく質を発現するため、蛍光顕微鏡を用いて遺伝子導入細胞を確認することができる(右パネル)。
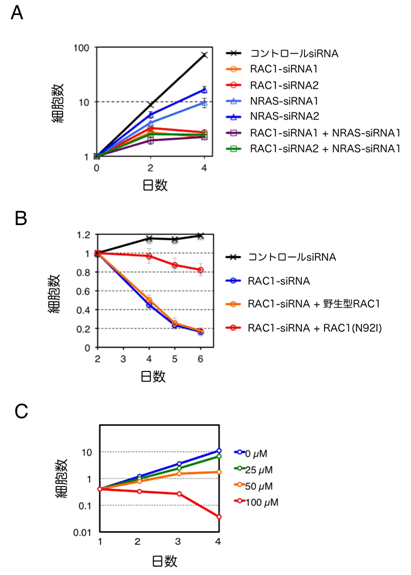
図3 RAC1がん遺伝子は新しい治療標的
- (A)遺伝子特異的なsiRNAを導入すると、特定の遺伝子の発現を抑制できる。ヒト線維肉腫細胞株HT1080に対してコントロールsiRNAを導入しても細胞増殖は抑制されないが、RAC1に対するsiRNA導入によって細胞死が誘導された。NRASに対するsiRNA導入は軽微な影響しかなく、RAC1とNRAS両者に対するsiRNA導入によってもRAC1に対するsiRNA単独導入時と差は認められない。
- (B)HT1080細胞にRAC1に対するsiRNAを導入すると著明な細胞死が誘導され、そこにさらに野生型RAC1を発現させても細胞増殖は抑制されたままだが、RAC1(N92I)を発現させると細胞増殖が回復する。
- (C)HT1080細胞培養液にRAC1活性阻害薬を添加すると、濃度依存的に細胞死が誘導された。

表1 ヒトのがん検体におけるRAC変異
今回のスクリーニングおよび公的データベースに登録された情報などから、RACファミリーには上記の変異がさまざまながん検体で存在することが明らかになった。
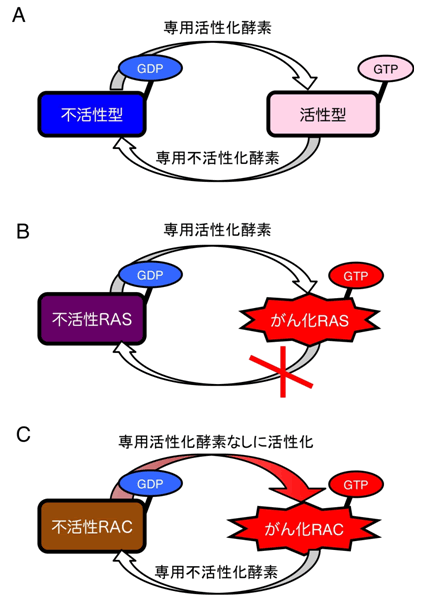
図4 変異RAS/RACの活性化機構
- (A)正常なRAS/RACファミリーたんぱく質は、一般にGDP結合状態が不活性型であり、GTP結合型が活性型である。両者間の移動はそれぞれ専用の酵素が触媒する。
- (B)アミノ酸置換によって活性化されたRASたんぱく質群は全て、結合したGTPが外れなくなった状態となり、その結果恒常的に活性化される。
- (C)一方今回発見した変異RAC群は、専用の酵素がなくてもGTP結合が自動的に生じる変異体であり、その結果恒常的に活性化される。
<用語解説>
- 注1) 横綱がん遺伝子
- ある正常な遺伝子が、異常をきたすことでがん化能を有するようになったものを、がん遺伝子といいます。その中で、強力ながん化能を有し、がん細胞の本質的な発がん原因である遺伝子を、本研究グループは「横綱がん遺伝子」と呼んでいます。横綱がん遺伝子の機能を抑制する薬剤は、がんに対する新たな特効薬になると考えています。
- 注2) EML4-ALK
- 本研究グループが発見した横綱がん遺伝子の1つ。肺がん細胞内において2番染色体内の小さな逆位が生じ、EML4遺伝子とALK遺伝子が融合してEML4-ALKがん遺伝子が生じます。EML4-ALK融合遺伝子は、本来細胞内骨格に結合するEML4たんぱく質のアミノ末端側約半分と、ALK受容体型チロシンキナーゼの細胞内領域とが融合した異常たんぱく質を産生します。EML4-ALK融合遺伝子は非小細胞肺がんの4~5%(50才以下の肺がんの約1/3)に生じることが知られています。
- 注3) 次世代DNAシーケンサー
- 核酸の塩基配列を決定する装置をDNAシーケンサーといいますが、一般に使用されているDNAシーケンサーは一度の実験で多くとも数百種類のDNA断片しか解析できません。しかし次世代DNAシーケンサーと呼ばれる装置は、一度の実験で数億から数10億種類ものDNA断片を同時に解析可能であり、大量の塩基配列情報を比較的簡便に得ることができ、ヒトゲノムの全体量より多い塩基配列データを取得可能です。
- 注4) 低分子量Gたんぱく質
- GTP(注6参照)に結合するたんぱく質を一般にGたんぱく質と呼びますが、なかでも分子量20~30kDa程度の小型たんぱく質群を低分子量Gたんぱく質と呼びます。ヒトゲノムには約150種類のメンバーがコードされています。
- 注5) GDP(guanosine diphosphate)
- グアノシン二リン酸。GTP(注6参照)の三リン酸からGTPアーゼによって1分子のリン酸が取り除かれたものです。
- 注6) GTP(guanosine triphosphate)
- グアノシン三リン酸。GDPがATPからリン酸を1分子受け取って生成されます。低分子量Gたんぱく質の多くはGDP結合型とGTP結合型の間を移り変わることで細胞内スイッチとして働きます。それ以外にも細胞内でGTPはエネルギー供与体として働き、また核酸の合成などにも利用されます。
- 注7) RASファミリー
- NRAS、HRAS、KRASからなるファミリーで、互いに極めて良く似た遺伝子です。いずれのメンバーもゲノム突然変異の結果、12番目、13番目あるいは61番目のアミノ酸置換を生じて、発がん能を獲得することが知られており、ヒトのさまざまながんで変異RASが広く認められます。
- 注8) siRNA(small interfering RNA)
- 21-23塩基からなる2本鎖RNAで、細胞内でダイサー・アルゴノートなど一連の酵素と反応した後、相補的な配列を持つmRNAに特異的に結合して分解します。人為的にsiRNAを合成することで、任意のmRNAを分解しノックダウンすることが可能です。
- 注9) トリプルネガティブ乳がん
- 乳がんの中には、エストロゲン受容体やプロゲステロン受容体などホルモン受容体が陽性の乳がんと、HER2遺伝子増幅が認められるHER2陽性乳がんがあり、それ以外にも、エストロゲン受容体、プロゲステロン受容体、HER2のいずれも陰性(トリプルネガティブ)な乳がんがあります。このタイプは乳がん全体の15~20%を占め、有効な分子標的治療があまりなく予後不良です。
<参考論文>
1)“Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer” Nature 448:561-566, 2007
doi: 10.1038/nature05945
2)“Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer” N Engl J Med 363:1693-1703, 2010
doi: 10.1056/NEJMoa1006448
<論文タイトル>
“ Transforming Mutations of RAC Guanosine Triphosphatases in Human Cancers”
(ヒト腫瘍におけるRAC GTP分解酵素のがん化変異)
doi: 10.1073/pnas.1216141110
<お問い合わせ先>
<研究に関すること>
間野 博行(マノ ヒロユキ)
自治医科大学 分子病態治療研究センター ゲノム機能研究部 教授
〒329-0498 栃木県下野市薬師寺3311-1
Tel:0285-58-7449 Fax:0285-44-7322
E-mail:
東京大学 大学院医学系研究科 ゲノム医学講座 特任教授
〒113-0033 東京都文京区本郷7-3-1
Tel:03-5841-0633 Fax:03-5841-0634
E-mail:
<JSTの事業に関すること>
原口 亮治(ハラグチ リョウジ)、井上 聡子(イノウエ アキコ)
科学技術振興機構 戦略研究推進部
〒102-0076 東京都千代田区五番町7 K’s五番町
Tel:03-3512-3525 Fax:03-3222-2067
E-mail: