JST 課題解決型基礎研究の一環として、自治医科大学 分子病態治療研究センターの間野 博行 教授(兼 東京大学 大学院医学系研究科 特任教授)らは、チロシンキナーゼ注1)阻害剤を用いた肺がん治療において、がんが薬剤耐性となる分子メカニズムを発見しました。
肺がんは先進国で最も死亡者数の多いがんで、有効な治療法の開発が待たれています。間野教授らは2007年に肺腺がんの細胞から新たな肺がんの原因遺伝子EML4-ALK注2)を発見しました。この遺伝子によって作られるEML4-ALKたんぱく質が、染色体転座の結果生じた活性型融合チロシンキナーゼ(以後、「EML4-ALKキナーゼ」)です。その酵素活性を抑制するALK阻害剤がEML4-ALK陽性肺がん(以後、「ALK肺がん」)患者の分子標的治療法として開発中であり、有効性が確認されています。しかし、一部の症例はALK阻害剤に対する薬剤耐性となっており、その原因解明が課題でした。
本研究グループは今回、当初は有効であったALK阻害剤が5ヵ月間の治療後に突然効かなくなった症例を経験し、その治療前と再発後のがん細胞を次世代DNAシークエンサー注3)で配列比較を行うことで、再発後にのみEML4-ALKキナーゼ内に新たな付加変異が2種類生じていることを発見しました。これら2種類の付加変異はどちらもEML4-ALKキナーゼを薬剤耐性型に変化させていることが明瞭に確認され、本症例の薬剤耐性の原因であると考えられます。
本研究で発見された付加変異を有するEML4-ALKキナーゼに対しても結合・阻害しうる構造を持ったALK阻害剤を開発すれば、今後の肺がん治療において薬剤耐性を誘導しにくい分子標的治療法が実現すると考えられます。
本研究成果は、2010年10月28日(米国東部時間)発行の米国医学雑誌「The New England Journal of Medicine」に掲載されます。
本成果は、以下の事業・研究領域・研究課題によって得られました。
戦略的創造研究推進事業 研究加速課題
| 研究課題名 |
: |
新規がん遺伝子同定プロジェクト
|
| 研究者名 |
: |
間野 博行
(自治医科大学 分子病態治療研究センター 教授/東京大学 大学院医学系研究科 特任教授)
|
| 研究期間 |
: |
平成20年1月~平成26年3月 |
上記研究課題では、研究代表者らが開発した遺伝子機能スクリーニング法と次世代DNAシークエンサーを用いたゲノミクス解析法とを組み合わせることで、がん臨床検体から発がん原因遺伝子を同定し、その知見に基づく新たな分子診断法・分子標的療法を開発することを目指しています。
<研究の背景と経緯>
たんぱく質のチロシン残基をリン酸化する酵素であるチロシンキナーゼは私たちの細胞の増殖を制御する重要な分子ですが、点突然変異や遺伝子融合などさまざまなゲノム変異によって恒常的活性型となることで発がんを誘導します。
肺がんは先進国におけるがん死因の第1位を占める予後不良の疾患です。本研究グループは2007年に肺がんの一部で2番染色体内の微小な転座が生じた結果、EML4-ALK融合がん遺伝子が生じることを発見しました(図1、参考論文1)。ALKは受容体型チロシンキナーゼですが、その酵素領域がEML4と融合することで酵素活性が上昇し強力ながん化能を獲得したEML4-ALKキナーゼになるのです。さらに本研究グループは2008年に、EML4-ALKキナーゼを肺特異的に産生する遺伝子改変マウスを作成し、このマウスが生後すぐに両肺に数百個もの肺腺がんを多発すること、またそのようなマウスにALK阻害剤を投与すると肺がんが消失することも報告しました(参考論文2)。これらの発見を受け、実際の肺がん患者に投与するALK阻害剤の開発が多くの製薬会社で行われており、すでに1社は独自のALK阻害剤(crizotinib注4))による第一相・第二相臨床試験を終了し、その成果を2010年の米国臨床腫瘍学会総会で発表しました(参考論文3)。それによるとcrizotinibの投与により、約9割の症例で腫瘍サイズがコントロールされ、完全寛解症例(腫瘍が消失する)まで出現するという驚くべきものでした。しかし一部の症例はcrizotinibが効きませんでした。このようなALK阻害剤耐性の原因は全く不明でした。
本研究グループは今回、ALK阻害剤が著効したものの半年後に突然再発した症例のがん細胞を解析することで、薬剤耐性メカニズムを明らかにすることに成功しました。
<研究の内容>
2010年春まで日本においてはALK阻害剤による臨床試験が行われてこなかったため、これに先立ち本研究グループは2009年春より、日本のALK肺がん患者を救うべくボランティアのEML4-ALK診断ネットワーク事業を行ってきました。本活動によりEML4-ALK陽性と診断された患者の一部は、海外で行われるALK阻害剤の臨床試験に参加して治療を受けてきました。そのなかに、ALK阻害剤治療によって当初腫瘍の著しい縮小や呼吸困難などの臨床症状の消失をみましたが、約5ヵ月間の治療後に突然再発し、薬剤耐性肺がんへと移行した患者がいました。
そこで、本症例で生じた薬剤耐性メカニズムを明らかにするべく、ALK阻害剤による治療前と再発後の患者がん細胞の比較を試みました。具体的な解析試料は前者が喀痰(かくたん)、後者が胸水であり、それぞれの試料内にがん細胞がどの程度の割合存在するかは全く不明です。そこで微量のがん細胞しか含まれていない試料からでも遺伝子変異を高感度に検出できるように、次世代DNAシークエンサーを用いた解析を行いました。EML4-ALKの酵素活性領域に新たな付加変異が生じている可能性を検証するため、ALK酵素活性領域のcDNAを次世代DNAシークエンサーによって解析し、治療前と再発後での変化を調べました。その結果、再発後にのみ新たに2種類の塩基置換(4374番目の塩基のG→A置換と4493番目の塩基のC→A置換)がALK酵素活性領域に出現していることを発見しました(図2A)。前者の塩基置換はEML4-ALKキナーゼの1156番目のアミノ酸であるシステイン(C)をチロシン(Y)に置換し、後者の塩基置換は1196番目のアミノ酸であるロイシン(L)をメチオニン(M)に置換します(図2B)。
さらに興味深いことに、これら2種類の変異は別々のcDNA上に生じており、この結果からC1156Y変異を有するがん細胞クローンとL1196M変異を有するクローンは患者腫瘍組織内において互いに独立に出現していることが明らかになりました。
次に、この2種類のアミノ酸変異が薬剤耐性の原因であるのかをマウス細胞で調べました。すると、EML4-ALKキナーゼを発現するマウスB細胞株BA/F3は、ALK阻害剤(crizotinib)を投与すると速やかに細胞死が誘導されましたが、EML4-ALK(C1156Y)やEML4-ALK(L1196M)キナーゼを発現するBA/F3細胞は、はるかに高濃度のcrizotinibを投与しなければ死滅しません(図3上)。すなわち、C1156YおよびL1196M変異のどちらも薬剤耐性を付与することが確認されました。しかも、本変異はcrizotinib治療中に生じたものであるにもかかわらず、別のALK阻害剤である2,4‐pyrimidinediamine derivative(PDD)に対しても、同様に耐性を獲得することが示されました(図3下)。従って本研究グループが発見した薬剤耐性変異は、広くALK阻害剤一般に対して耐性原因となる遺伝子変異であると予想されます。
このC1156YやL1196M変異がどのようなメカニズムで薬剤耐性をもたらすかを検討するため、ALK酵素活性領域の立体構造モデル上にC1156とL1196部位を示したのが図4です。チロシンキナーゼは上下の2種類の丸い構造体が中央の“ちょうつがい”でつながっているような構造をしており、その“ちょうつがい”部分に酵素活性の補酵素であるATP(adenosine triphosphate)注5)が結合します。またcrizotinib、PDDを含む多くのキナーゼ阻害剤は、このATP結合ポケットにATPと競合的に結合し、酵素のATPとの結合と活性化をブロックします。C1156はATP結合ポケットの上端に位置しており直接ATPとは接していないと予想されますので(図4左)、この部位がチロシンに変化することで酵素上部の変形が生じ阻害剤の挿入が不可能になったのではないかと予想されます。一方L1196部位はATP結合ポケットの最深部に位置しますので(図4右)、この部位が、大きな側鎖を持つアミノ酸であるメチオニンに置換されることでALK阻害剤が結合できなくなったと考えられます。
興味深いことにこのL1196部位は、他のがん化チロシンキナーゼが薬剤耐性となる際に変異する場所と同じでした。肺がんの一部はEGFR注6)チロシンキナーゼの活性型異常によって生じると考えられますが、そのような症例にはEGFR阻害剤(gefitinib/erlotinib)が有効であることが知られています。しかし、EGFR阻害剤の治療中に一部の症例が薬剤耐性となることがあり、その細胞の解析から790番目のスレオニン(T)がメチオニン(M)へ変化することが薬剤耐性の原因であることが明らかになっています(図5)。また、慢性骨髄性白血病はABLチロシンキナーゼが染色体転座の結果BCRと融合してBCR-ABL注7)融合チロシンキナーゼが生じることにより発症しますが、そのような症例にはABLの酵素活性阻害剤であるimatinibが有効です。しかしimatinib治療中に薬剤耐性となる症例が出現することがあり、ABL内の315番目のアミノ酸であるスレオニン(T)がイソロイシン(I)へと変化する異常が最も重要な薬剤耐性原因であることが分かっています(図5)。EGFRのT790もABLのT315もどちらもATP結合ポケットの最深部に位置しており、この場所は「gate keeper(守衛)部位」と呼ばれています。今回の発見で、EGFRやABLとは全く異なるキナーゼ(ALK)を全く異なる阻害剤で治療したにもかかわらず、同じgate keeper部位の変異が薬剤耐性をもたらすことが分かりました。
<今後の展開>
今回の発見により、ALK阻害剤の耐性メカニズムが解明され、チロシンキナーゼ阻害剤耐性メカニズムに共通性があることが明らかになりした。これは今後のチロシンキナーゼ阻害剤の開発に大きな影響を与えると考えられます。まずALK肺がんに対する治療だけに限っても、これ以降の新たなALK阻害剤開発の際にはC1156Y変異やL1196M変異があっても有効にALK阻害活性を発揮しうる(つまりALKのATP結合部位に結合しうる)構造を有するALK阻害剤を開発すればよいことになります。
しかも今回の発見は、チロシンキナーゼ阻害剤に対する耐性メカニズムとしてgate keeper部位の異常が、チロシンキナーゼ・阻害剤の種類にかかわらず広く共通の原因となることを示唆しています。従って、どのような酵素を対象とした場合でも、そのATP結合部位に競合的に結合する阻害剤を開発する場合は、gate keeper部位に大きな側鎖を持つアミノ酸が挿入されたとしても阻害活性を失わないような化合物を最初からデザインするべきであるという指針が示され、今後の薬剤耐性がん治療への道が開かれました。
<参考図>
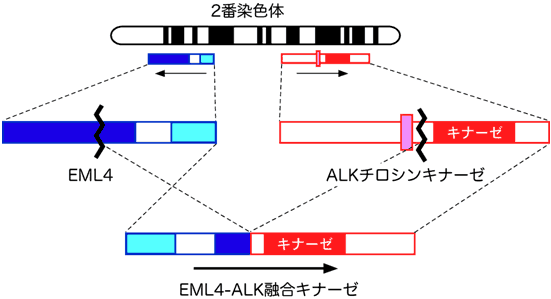
図1 EML4-ALK融合遺伝子
EML4遺伝子とALK遺伝子は、正常細胞においてどちらもヒト2番染色体上のごく近い位置に互いに反対向きに存在するが、両遺伝子を挟む領域が逆位となることでEML4-ALK融合遺伝子が生じる。その結果微小管会合たんぱくEML4のアミノ末端側約半分と受容体チロシンキナーゼALKの細胞内キナーゼ領域とが融合した活性型融合キナーゼであるEML4-ALKキナーゼが肺がんの中で産生されることになる。
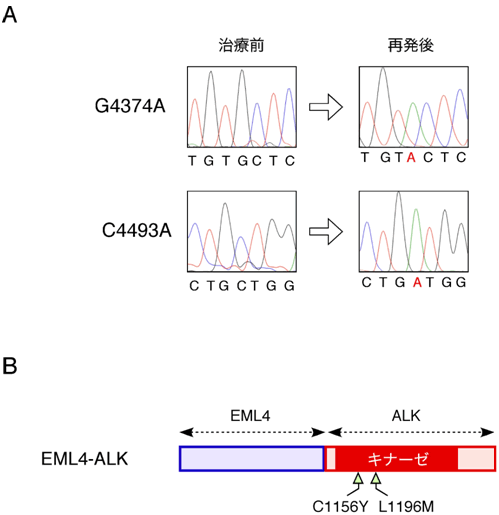
図2 薬剤耐性期に生じた2種類のアミノ酸置換
- (A) 治療前と再発後のがん検体からALK cDNAを調整して次世代DNAシークエンサーで解析したところ、2種類の塩基置換が再発後のみ存在することが分かった。本塩基置換を一般的なDNAシークエンサーで確認した図を示す。4374番目の塩基であるグアニン(G)と4493番目の塩基であるシトシン(C)が、それぞれアデニン(A)塩基に置き換わっていることが分かる。
- (B) G4374A変化の結果、1156番目のアミノ酸であるシステイン(C)がチロシン(Y)残基に変化し、一方C4493A変化の結果、1196番目のロイシン(L)がメチオニン(M)残基に置き換わる。
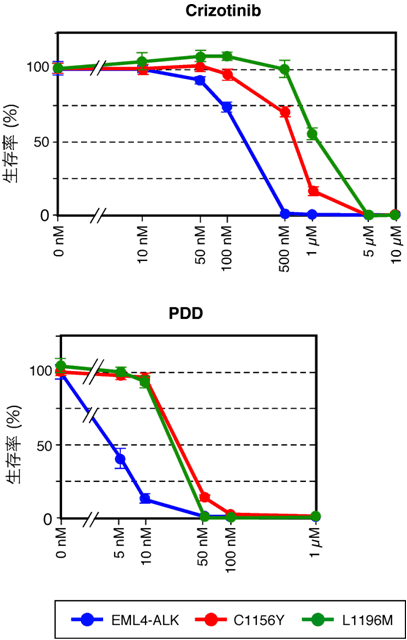
図3 C1156YとL1196M変異は、いずれも薬剤耐性の原因である
EML4-ALKキナーゼを発現させたBA/F3細胞の培養上清に、治療に用いたALK阻害剤(crizotinib)を添加するとその濃度依存性に細胞死が誘導される(青色)。しかしC1156Y(赤色)あるいはL1196M(緑色)変異を持つEML4-ALKキナーゼの場合は、明瞭にcrizotinibに対する感受性が低下した(上図)。一方、治療には用いられなかった別のALK阻害剤(PDD)の場合も同様に感受性低下が確認された(下図)。
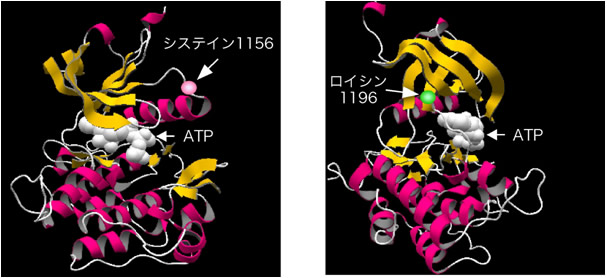
図4 ALK酵素活性領域の立体構造
ALK酵素活性領域の3次元構造を、類似のチロシンキナーゼであるINSRの構造(エントリーID“1ir3”、日本蛋白質構造データバンク(PDBj:Protein Data Bank Japan)のPDB検索からエントリーIDを入力すると情報が得られます。PDBjホームページ http://www.pdbj.org/index_j.html)をもとに類推した。αヘリックスおよびβシートをそれぞれピンクおよびオレンジ色で表した。ALK酵素活性領域は、ATP結合ポケットを中心に上部と下部の大きく2種類の領域からなることが分かる。1156番目のシステイン残基はATP結合ポケットの上端に位置している(左図)。一方左図の構造を左横から見た図を右に示した。1196番目のロイシン残基はATP結合ポケットの一番奥に位置していることが分かる。
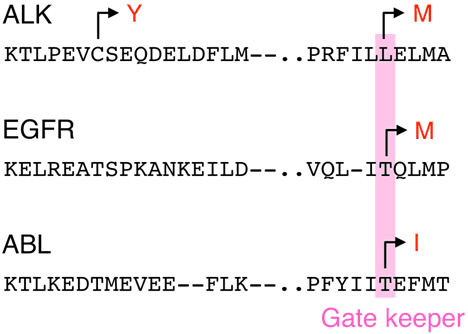
図5 他の活性型チロシンキナーゼにおける薬剤耐性変異
現在広くがん治療に用いられている標的チロシンキナーゼであるEGFR(肺がん)とABL(慢性骨髄性白血病)について、そのアミノ酸配列をALKの配列と並べて示した。
活性型EGFRに対して用いられるgefitinibに耐性となる変異で一番多い例は790番目のスレオニン残基(T)がメチオニン(M)へ変化する場合であり、BCR-ABLに対して用いられるimatinibに耐性となる変異の最も多い例は315番目のスレオニン(T)がイソロイシン(I)となる場合である。EGFRのT790とABLのT315はどちらもたんぱく構造上同じ位置に存在しており(ATP結合ポケットの最深部)、gate keeperポジションと名付けられている。興味深いことに、今回EML4-ALKキナーゼで見つかった変異部位であるL1196もこのgate keeperポジションに位置していた。一方EML4-ALKキナーゼのC1156部位は他のキナーゼの耐性変異として報告されていない。
<用語解説>
- 注1) チロシンキナーゼ
- キナーゼとは基質をリン酸化する酵素の総称であり、そのうちチロシンキナーゼは基質たんぱくのチロシン残基をリン酸化する酵素のこと。一般にその活性化は私たちの細胞の増殖を正に誘導します。
- 注2) EML4-ALK
- 肺がん細胞内において2番染色体内の小さな逆位が生じ、EML4遺伝子とALK遺伝子が融合してEML4-ALKがん遺伝子が生じます。EML4-ALK融合遺伝子は、本来細胞内骨格に結合するEML4たんぱく質のアミノ末端側約半分と、ALK受容体型チロシンキナーゼの細胞内領域とが融合した異常たんぱく質を産生します。EML4-ALK融合遺伝子は非小細胞肺がんの4~5%(50才以下の肺がんの35%)に生じることが知られています。
- 注3) 次世代DNAシークエンサー
- 核酸の塩基配列を決定する装置をDNAシークエンサーと言いますが、一般に使用されているDNAシークエンサーは一度の実験で多くとも数百種類のDNA断片しか解析できません。しかし次世代DNAシークエンサーと呼ばれる装置は、一度の実験で数百万から数億種類ものDNA断片を同時に解析可能であり、大量の塩基配列情報を比較的簡便に得ることができます。現在、3種類ほどの次世代DNAシークエンサーが入手可能です。
- 注4) crizotinib
- ALKチロシンキナーゼとMETチロシンキナーゼの酵素活性を選択的に阻害する薬剤で、現在ALK肺がんに対する国際第三相臨床試験が行われています。
- 注5) ATP(adenosine triphosphate)
- アデノシン三リン酸。細胞内のさまざまな化学反応において広く利用される補酵素で、ADP+Piに分解される際に放出する高エネルギーが多くの化学反応に利用されます。チロシンキナーゼはATPの分解エネルギーを利用するだけでなく、そこで生じるリン酸を直接基質のリン酸化反応に利用しています。
- 注6) EGFR(Epidermal Growth Factor Receptor)
- 皮膚など上皮系の細胞に働いて、細胞増殖を促すたんぱく質を上皮成長因子と言いますが、その細胞側の受容体がEGFRです。EGFRたんぱく質は細胞内領域にチロシンキナーゼ酵素活性を有しており、上皮成長因子が結合すると、そのキナーゼ活性が誘導されます。一部の肺がん症例において、EGFRの遺伝子異常が見つかりました。この異常はEGFRたんぱく質のキナーゼ活性を高め、上皮成長因子が結合していない状態でも、EGFR酵素活性を上昇させて肺がん発症を誘導するとされています。
- 注7) BCR-ABL
- 慢性骨髄性白血病細胞において、9番染色体と22番染色体との間で転座が生じ、前者に存在するABL遺伝子と後者に存在するBCR遺伝子が融合したBCR-ABL遺伝子が産生されます。ABLたんぱくはチロシンキナーゼとしての活性を有していますが、BCRと融合することで酵素活性が上昇し発がん能力を獲得します。なお正常BCRの機能は現在でもよく分かっていません。
<参考論文>
- 1) “Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer” Nature 448:561-566, 2007
- 2) “A mouse model for EML4-ALK-positive lung cancer” PNAS 105:19893-19897, 2008
- 3) “Clinical activity of the oral ALK inhibitor PF-02341066 in ALK-positive patients with non-small cell lung cancer (NSCLC)” J Clin Oncol 28:18s (supple; abstr 3), 2010
<論文名>
“Mutations of EML4-ALK in Lung Cancer That Confer Resistance to ALK Inhibitors”
(肺がんにおいてALK阻害剤の耐性原因となるEML4-ALKキナーゼ内付加変異の発見)
<お問い合わせ先>
<研究に関すること>
間野 博行(マノ ヒロユキ)
自治医科大学 分子病態治療研究センター ゲノム機能研究部 教授
〒329-0498 栃木県下野市薬師寺3311-1
Tel:0285-58-7449 Fax:0285-44-7322
E-mail: 東京大学 大学院医学系研究科 ゲノム医学講座 特任教授
東京大学 大学院医学系研究科 ゲノム医学講座 特任教授
〒113-0033 東京都文京区本郷7-3-1
Tel:03-5841-0633 Fax:03-5841-0634
E-mail:
<JSTの事業に関すること>
古旗 憲一(フルハタ ケンイチ)
独立行政法人 科学技術振興機構 イノベーション推進本部 研究推進部
〒102-0075 東京都千代田区三番町5 三番町ビル
Tel:03-3512-3526 Fax:03-3222-2065
E-mail: