ポイント
- 損傷ミトコンドリアを分解する品質管理システム(マイトファジー)は重要な生体防御機構。
- ミトコンドリアに存在するアポトーシス抑制たんぱく質は、ミトコンドリアと一緒に分解されないことを発見。
- パーキンソン病などマイトファジーの異常が原因と考えられる病気の解明に期待。
JST 課題達成型基礎研究の一環として、九州大学 生体防御医学研究所の白根 道子 准教授、中山 敬一 教授らは、細胞の品質管理システムの1つで、損傷したミトコンドリアを除去する「マイトファジー(自食作用)注1)」のメカニズムの一部を解明しました。
ミトコンドリアは、エネルギー産生を担う重要な細胞内小器官ですが、同時に有害な活性酸素も産出するため、損傷したミトコンドリアの蓄積は細胞に悪影響を及ぼします。そのため、正常な細胞では損傷したミトコンドリアだけを選択的に除去するマイトファジーというシステムが働いていますが、ミトコンドリアが分解されるとミトコンドリアに存在するアポトーシス(細胞死)注2)抑制たんぱく質まで消失するため、過剰にアポトーシスが起きる危険があります。細胞がマイトファジーの際にどのようにしてこの危険を回避しているかは今まで謎でした。
本研究チームは、マウスの細胞を用いてマイトファジーで損傷したミトコンドリアを分解させた際に、ミトコンドリアに存在したアポトーシス抑制たんぱく質「FKBP38注3)」が速やかに小胞体へと避難し、分解から免れることを見いだしました。小胞体へ避難したFKBP38がアポトーシスを抑制することにより、細胞はマイトファジーの際にもアポトーシスを起こさずに生存できることが分かりました。FKBP38を持たせないマウスの細胞でマイトファジーを起こさせると、FKBP38を持つ正常の細胞に比べてアポトーシスが多く起こっていました。
近年、マイトファジーはパーキンソン病注4)の原因遺伝子の1つであるParkin注5)によって制御されていることが明らかにされ、マイトファジーの異常による損傷ミトコンドリアの蓄積がパーキンソン病に関与すると報告されています。今後さらにマイトファジーの仕組みが解明されることで、パーキンソン病などマイトファジーが関与すると考えられる病気の新規治療法確立が期待されます。
本研究成果は、2013年1月29日(英国時間)に英国科学雑誌「Nature Communications」のオンライン版で公開されます。
本成果は、以下の事業・研究領域・研究課題によって得られました。
戦略的創造研究推進事業 チーム型研究(CREST)
| 研究領域 |
「生命システムの動作原理と基盤技術」
(研究総括:中西 重忠 (公財)大阪バイオサイエンス研究所 所長)
|
| 研究課題名 |
「ユビキチンシステムの網羅的解析基盤の創出」 |
| 研究代表者 |
中山 敬一(九州大学 生体防御医学研究所 教授) |
| 研究期間 |
平成19年10月~平成25年3月 |
JSTはこの領域で、生命システムの動作原理の解明を目指して、新しい視点に立った解析基盤技術を創出し、生体の多様な機能分子の相互作用と作用機序を統合的に解析して、動的な生体情報の発現における基本原理の理解を目標としています。上記研究課題では、細胞分裂、DNA修飾、たんぱく質の品質管理など重要な生命現象を調節するユビキチンシステムについて、遺伝学とプロテオミクスを組み合わせた新しい方法によって網羅的に解析し、システムの全体像を解明することを目指しています。
<研究の背景と経緯>
ミトコンドリアは、細胞内エネルギーを作り出す重要な細胞小器官でありながら、細胞死(アポトーシス)でも中心的な役割を担い、細胞の生と死の両方の制御に非常に重要な役割を果たしています。アポトーシスは、個体をより良い状態に保つために、細胞が自殺するプログラムされた細胞死で、そのため厳密にコントロールされています。特に、ミトコンドリアの中にあるFKBP38などの抗アポトーシスたんぱく質ファミリーが、無意味にアポトーシスが起こらないように積極的に抑制しています。
ミトコンドリアがエネルギーを作り出す際には、同時に活性酸素が作り出されるため、徐々にミトコンドリアが傷害され機能が低下していきます。損傷したミトコンドリアの蓄積は細胞全体に悪影響を及ぼすため、機能低下したミトコンドリアはマイトファジー(自食作用)というシステムで選択的に分解・除去されます(図1)。近年、このマイトファジーの異常で、損傷したミトコンドリアが細胞内に蓄積することが、パーキンソン病発症の一因(ミトコンドリア機能障害仮説注6))ではないかと考えられるようになりました。
このように、細胞の健康にとって重要なマイトファジーですが、ミトコンドリアが分解されるとミトコンドリアに存在しているアポトーシスを抑制するたんぱく質まで消失するため、過剰にアポトーシスが起きる危険があります。細胞が、マイトファジーを起こした際にどのようにしてこの危険を回避しているかは今まで謎でした。
<研究の内容>
本研究チームは、CRESTの研究目的である「たんぱく質の網羅的解析技術」を用いて、ミトコンドリア機能に関わる分子の機能について研究を行いました。特にアポトーシスの制御に関わるFKBP38というたんぱく質に注目し、マイトファジーとアポトーシスの関連性を調べました。まず、マウスの線維芽細胞でマイトファジーを起こさせたところ、ほとんど全てのミトコンドリア自体は分解されましたが、そこに存在するFKBP38は分解されずにミトコンドリアから小胞体へと移動していました(図2)。このことから、FKBP38が小胞体上で何らかの機能を果たしていることが推測されました。
次に、小胞体上に移動したFKBP38がアポトーシスの抑制に働いているかを検証するために、FKBP38を人工的に持たせないマウス(ノックアウトマウス)から作製した線維芽細胞を用いて、マイトファジーを起こさせたところ、FKBP38を持たない細胞ではアポトーシスが多く起こっていました(図3)。よって、マイトファジーの際に分解を免れたFKBP38は、避難した小胞体上でアポトーシス抑制に働いていることが分かりました(図4)。さらに、ミトコンドリアに存在するほかの抗アポトーシスたんぱく質についても検討したところ、抗アポトーシスたんぱく質Bcl-2注7)もFKBP38と同様に、マイトファジーの際にミトコンドリアから小胞体へと避難することが分かりました。
そのミトコンドリアから小胞体への移動の機構を詳細に調べたところ、FKBP38のアミノ酸配列中に、細胞内でのFKBP38の居場所の変化を決めている重要な配列を発見しました。マイトファジーの際に居場所が変化するFKBP38とBcl-2の配列を、変化しない別のミトコンドリアたんぱく質の配列と比較したところ、C末端領域の塩基性アミノ酸注8)の数が少ないことが分かりました。そこで、アミノ酸置換により塩基性度を上げたFKBP38とBcl-2の変異体を作製してマイトファジーを誘導すると、FKBP38とBcl-2はミトコンドリアから小胞体へと避難できなくなることが分かりました(図5)。
本研究より、抗アポトーシスたんぱく質のFKBP38は、マイトファジーの際ミトコンドリアから小胞体へと避難することで自らの分解を免れ、アポトーシス抑制に働くことを見いだしました。またその居場所の変化には、FKBP38のC末端領域のアミノ酸の塩基性度が重要であることが明らかになりました。
<今後の展開と応用への期待>
本研究は、マイトファジーが起きた際にミトコンドリアの抗アポトーシスたんぱく質が小胞体へ避難して分解を逃れ、引き続きアポトーシスを抑制することを示した初めての報告です。近年、マイトファジーの不全による損傷ミトコンドリアの蓄積がパーキンソン病の発症に関与するのではないかと示唆されています。そのため、マイトファジーの機構を解明することは、細胞生物学的な知見の蓄積に加え、マイトファジーの異常が原因となる神経疾患の克服といった面からも強く期待されています。また、今後、FKBP38の機能を制御しアポトーシスを調節することによりドーパミンを放出する神経細胞の変性(細胞死)を阻止するなど、パーキンソン病治療への応用も期待されます。
<参考図>
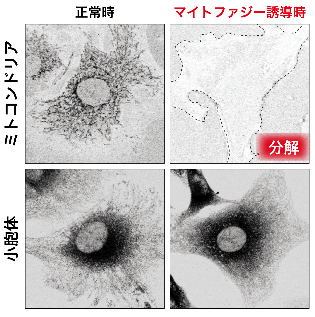
図1 マイトファジーによる損傷ミトコンドリアの選択的な分解
マウス初代繊維芽細胞において人工的にミトコンドリアに損傷を与えると、小胞体は分解されず残るのに対し(下段右)、ミトコンドリアは選択的に分解除去されます(上段右)。
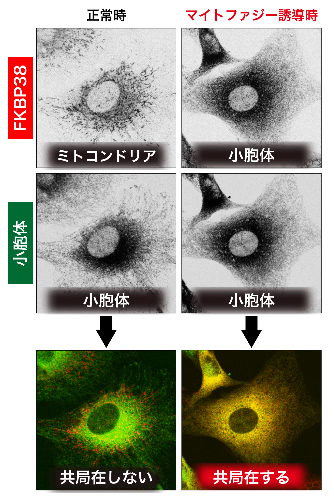
図2 マイトファジーを起こした際のFKBP38の局在移動
FKBP38は正常時にはミトコンドリアに局在していますが(上段左)、マイトファジー誘導時には小胞体へと局在変化します(上段右)。FKBP38(上段、赤色)と小胞体(中段緑色)が一致すると、重ね合わせ画像(下段)で黄色に表示されます。
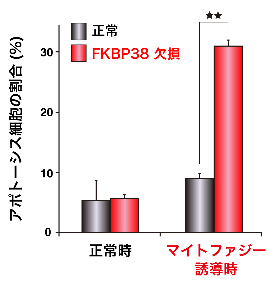
図3 FKBP38を持たない細胞におけるマイトファジー誘導時のアポトーシス
マイトファジー誘導時に起こるアポトーシスの割合は、FKBP38を持たない細胞(赤)では正常細胞(黒)に比べて約3倍増加します。
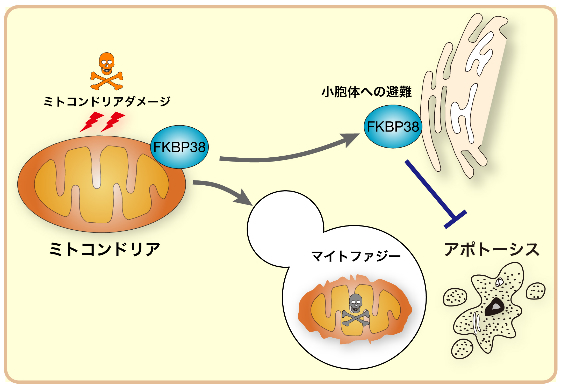
図4 FKBP38によるマイトファジー時のアポトーシス抑制機構
ミトコンドリアが傷害を受けると、マイトファジーのシステムが働き、損傷ミトコンドリアの分解が起こります。その際、FKBP38はミトコンドリアから小胞体へと避難するために分解から免れ、小胞体上でアポトーシス抑制に働くことが明らかとなりました。
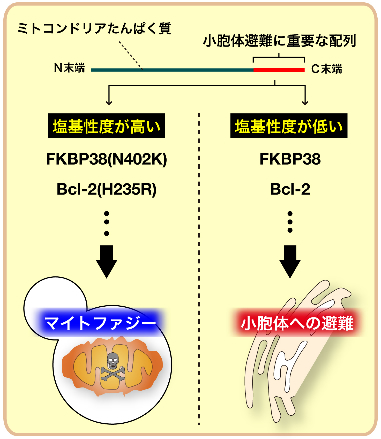
図5 マイトファジーの際、ミトコンドリアから小胞体への避難に重要な配列
ミトコンドリアたんぱく質のC末端領域の配列が、マイトファジーが起きた際の小胞体への避難の有無を決定しています。すなわちC末端領域中の塩基性アミノ酸の数と塩基性度が重要で、FKBP38やBcl-2は塩基性度が低くマイトファジーの際にミトコンドリアから小胞体へと避難します(右)。一方、塩基性アミノ酸を変異させて塩基性度を高くしたFKBP38(N402K)やBcl-2(H235R)は、マイトファジーの際に避難できず、ミトコンドリアに局在したままで分解されます(左)。
<用語解説>
- 注1) マイトファジー(自食作用)
- ミトコンドリアのオートファジー(自食作用)をマイトファジーといい、ミトコンドリアを選択的に分解し細胞内を浄化する、細胞の品質管理システムの1つです。ミトコンドリアはエネルギー産生を担う重要な細胞内小器官ですが、同時に有害な活性酸素も産出するため、損傷ミトコンドリアの蓄積は細胞に悪影響を及ぼします。そのため機能不全となったミトコンドリアはマイトファジーにより除去されます。2008年にパーキンソン病関連因子のParkin注5)がマイトファジーで重要な働きをしていることが報告され、マイトファジーとパーキンソン病注4)との関係が注目されるようになりました。
- 注2) アポトーシス(細胞死)
- アポトーシスとは細胞が必要に応じて自殺するプログラムされた細胞死で、ミトコンドリア膜の崩壊を介して死に至るミトコンドリアに依存した経路と、ミトコンドリアに依存しない経路とがあります。FKBP38注3)やBcl-2注7)はミトコンドリアに依存したアポトーシスを抑制する抗アポトーシスたんぱく質で、ミトコンドリア膜上に存在しています。無秩序にアポトーシスが起きることは生物にとって非常に危険なため、抗アポトーシスたんぱく質によって細胞は守られています。
- 注3) FKBP38
- FKBP38はもともと神経線維(神経細胞の細胞体から延びる細長い突起)の腫瘍で活発に作り出されるたんぱく質として報告されていましたが、2003年に本研究チームにより、FKBP38はBcl-2注7)をミトコンドリアに局在させ、アポトーシス抑制に働いていることが発見されました。
- 注4) パーキンソン病
- 動作緩慢(寡動)・手足の震え(振戦)・筋肉のこわばり(固縮)などの運動障害を主徴とする神経疾患です。患者数の割合は人口10万人当たり約100人で、多くは初老期に発症しますが一部で若年に発症します。神経細胞(特に中脳にある黒質のドーパミンを産生する細胞)が死ぬことにより神経伝達物質のドーパミンが減少し、運動の制御に関わる黒質の線条体系が機能しなくなります。治療法としてはドーパミン補充療法(レボドパ治療)が基本ですが、副作用の問題がありドーパミンを受け取り、情報を伝達する受容体を刺激する薬やドーパミンの分解を抑制する薬と併用されます。いずれも対症療法で、原因となる神経細胞の死を阻止する根本的な治療法の開発が期待されています。
- 注5) Parkin
- Parkin遺伝子の変異は若年性パーキンソン病の原因として最も多く報告されています。Parkinはユビキチン化酵素の1つで、ユビキチンというたんぱく質の目印を付けて基質の分解を導きます。Parkinが損傷ミトコンドリア上のたんぱく質にユビキチンを付加し、ミトコンドリアの分解・除去を誘導していると考えられています。Parkinに変異があるパーキンソン病患者では、分解の目印が付かないために損傷ミトコンドリアが蓄積し神経細胞が変性すると考えられていますが、詳細な機構はいまだ不明です。別名「PARK2」とも呼ばれます。
- 注6) ミトコンドリア機能障害仮説
- パーキンソン病患者において、ミトコンドリア機能の低下が観察されています。また、除草剤や殺虫剤などに含まれるMPTPやロテノンなどのミトコンドリア機能を傷害する化学物質にさらされることにより、人や実験動物においてパーキンソン病に類似した行動の障害と神経細胞の変化が起こります。さらに1990年代に入って家族性パーキンソン病の原因遺伝子が次々と報告されるようになりましたが、最近になってそれらの多くがミトコンドリア機能に関与していることが明らかにされました。これらの事実から、パーキンソン病の原因がミトコンドリア機能の障害であることが提唱されています。
- 注7) Bcl-2
- 悪性リンパ腫細胞から発見されたBcl-2は、アポトーシスを強力に抑える抗アポトーシスたんぱく質です。Bcl-2は細胞の長期生存に必要ですが、この分子が過剰に働くとがんになります。
- 注8) 塩基性アミノ酸
- アミノ酸のうち、塩基性のアミノ基を持つものの総称です。たんぱく質は20種類のアミノ酸から構成されていますが、その中のリシン・アルギニン・ヒスチジンの3種類が塩基性アミノ酸です。
<論文名>
“Selective escape of proteins from the mitochondria during mitophagy”
(マイトファジー時におけるミトコンドリアからの選択的なたんぱく質の避難)
doi: 10.1038/ncomms2400
<お問い合わせ先>
<研究内容に関すること>
中山 敬一(ナカヤマ ケイイチ)
九州大学 生体防御医学研究所 分子医科学分野 教授
〒812-8582 福岡県福岡市東区馬出3-1-1
Tel:092-642-6815 Fax:092-642-6819
E-mail:
<JSTの事業に関すること>
石正 茂(イシマサ シゲル)
科学技術振興機構 戦略研究推進部 ライフイノベーショングループ
〒102-0076 東京都千代田区五番町7 K’s五番町
Tel:03-3512-3524 Fax:03-3222-2064
E-mail: