JST 課題達成型基礎研究の一環として、慶應義塾大学 医学部の永井 重徳 助教らは、自己免疫疾患の原因となる免疫細胞が増える、新たな免疫調節の仕組みを発見しました。
関節リウマチ、炎症性腸疾患注1)などの自己免疫疾患は、免疫システムが自分自身の正常な細胞や組織に対してまで攻撃してしまうため発症しますが、その原因として免疫システムで司令塔の役割をするヘルパーT細胞(T細胞の一種である細胞、以下、Th細胞)の細胞のなかでも、近年発見された「Th17細胞注2)」が大きく関与していると考えられています。そのため、自己免疫疾患の治療標的として世界中で盛んに研究されていますが、Th17細胞がどのように増えるのか、その仕組みは十分には明らかになっていません。
本研究グループは今回、脂質リン酸化酵素の一種である「PI3K注3)」がTh17細胞を増やす仕組みを明らかにし、さらにその仕組みを阻害する薬剤を自己免疫性腸炎のモデルマウスに投与して、症状を改善することに成功しました。
PI3KはTh17細胞を増やすだけでなく、さまざまな細胞で細胞分裂や代謝を起こす重要な役割を担っています。今回明らかになった新たな仕組みをさらに研究することによって、Th17細胞の増加にのみ関わるたんぱく質を抑制することができれば、さまざまな自己免疫疾患に対する、副作用の少ない治療法の開発につながるものと期待されます。
本研究成果は、2012年3月29日(米国東部時間)に米国オンライン科学雑誌「Cell Reports」で公開されます。
本成果は、以下の事業・研究領域・研究課題によって得られました。
戦略的創造研究推進事業 チーム型研究(CREST)
| 研究領域 |
「アレルギー疾患・自己免疫疾患などの発症機構と治療技術」
(研究総括:菅村 和夫 宮城県立病院機構 理事長) |
| 研究課題名 |
「細胞内シグナル制御による免疫リプログラミング」 |
| 研究代表者 |
吉村 昭彦(慶應義塾大学 医学部 微生物学・免疫学教室 教授) |
| 共同研究者 |
永井 重徳(慶應義塾大学 医学部 微生物学・免疫学教室 助教) |
| 研究期間 |
平成20年10月~平成26年3月 |
JSTはこの領域で、アレルギー疾患や自己免疫疾患を中心とするヒトの免疫疾患を予防・診断・治療することを目的に、免疫システムを適正に機能させる基盤技術の構築を目指しています。上記研究課題では、細胞内のシグナル伝達制御機構の解明とその人為的な調節により新たな免疫疾患治療の方法論を開発することを目指します。
<研究の背景と経緯>
生体の免疫システムでは、免疫細胞と呼ばれる細胞がさまざまな病原体を監視して私たちが病気にならないように働いています。免疫細胞の一種であるヘルパーT(Th)細胞は、病原体の種類に応じてTh1、Th2、Th17細胞に分化することが知られており、これらの細胞によって効率良く病原体が排除されます。Th17細胞は、炎症反応を引き起こして病原体を排除しますが、自己免疫疾患である関節リウマチや炎症性腸疾患などを悪化させる細胞であるとも考えられています。Th17細胞の分化の仕組みが分かれば、Th17細胞の増加が原因となる自己免疫疾患の治療につながる可能性があるため、その解明が期待されています。
未分化なT細胞がTh17細胞へと分化するためには、抗原による刺激とともにインターロイキン6やTGFβというたんぱく質(サイトカイン注4))が必要であり、これらの刺激によって発現するRORγという転写因子注5)がTh17細胞の分化に必須であることはすでに知られていました。一方、脂質リン酸化酵素の一種であるPI3K(以下、PI3K)はさまざまな細胞で細胞分裂や代謝などの細胞機能を調節する重要な役割を担っています。免疫細胞においても、PI3Kが免疫細胞の分化や機能に重要な役割を果たすことが知られており、Th17細胞の分化にも関与されていることが予想されていましたが、どのように関わっているかについては不明でした。
<研究の内容>
本研究グループはPI3KとTh17細胞の関係を調べるため、未分化のT細胞をTh17細胞に分化させる時にPI3Kの酵素活性を阻害する薬剤を加え、Th17細胞への分化が抑制されることを発見しました。また、PI3Kを破壊した未分化のT細胞をTh17細胞に分化させる実験も行い、同様に抑制されることを確認しました(図1A、B)。
次に、PI3Kとともに機能するたんぱく質に注目しました。PI3Kの活性化はAktというたんぱく質リン酸化酵素注6)の活性化を促し、AktはmTORC1注7)というたんぱく質複合体を活性化することが知られています。このAktの活性を人為的に増強させたところ、Th17細胞の分化が促進されました。また、mTORC1阻害剤(ラパマイシン注8))で処理した場合や、mTORC1の機能を破壊したT細胞でも、Th17細胞への分化が抑制されることを見いだしました(図1C)。さらに、実際に自己免疫性の大腸炎を発症するモデルマウスにラパマイシンを投与したところ、Th17細胞の分化誘導が抑制されるとともに、その症状が軽度になることが分かりました(図2)。すなわち、PI3K-Akt-mTORC1経路を抑制することによってTh17細胞の分化が抑制され、自己免疫性の炎症が抑制できることが分かりました。
また、このPI3K-Akt-mTORC1経路によって、どのようにTh17細胞の誘導が引き起こされるかについて、その仕組みも調べました。その結果、この経路はTh17細胞の増殖を抑える「Gfi-1」と呼ばれる転写因子を抑制することが分かりました(図3)。これに加え、Th17細胞の誘導に必須であることが知られていた転写因子「RORγ」を核に移動させ機能させるためには、PI3K-Akt-mTORC1経路が必要であることも明らかになりました(図4、図5)。
<今後の展開>
本研究によって、PI3K-Akt-mTORC1経路がTh17細胞の分化を促進する仕組みが解明され、この経路の阻害剤の投与で自己免疫疾患のモデルマウスの症状が実際に改善されました。しかし、このPI3K-Akt-mTORC1経路はほかのさまざまな細胞機能に関わるため、この経路を阻害して自己免疫疾患の症状を改善しようとする場合は、副作用を避ける工夫が必要です。
今回、Th17細胞への分化はPI3K-Akt-mTORC1経路によって、少なくとも2通りに制御されていることが明らかになりました。特にRORγを核に移動させることによってTh17細胞の分化を促進しているという発見は、ほかの細胞には影響を与えずにTh17細胞の分化を抑制できる免疫抑制剤の開発に貢献すると期待されます。
<参考図>
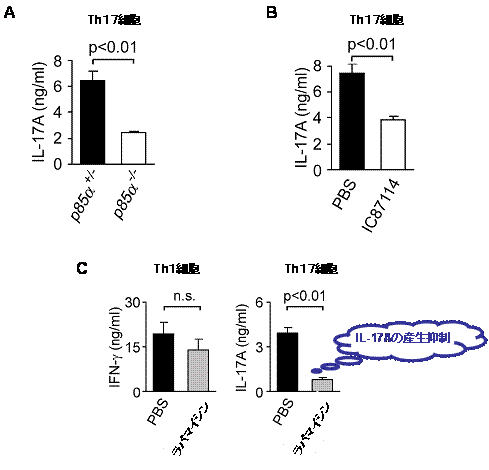
図1 PI3K-Akt-mTORC1経路はTh17細胞の分化を促進する
-
A:未分化なT細胞を、クラスⅠA PI3Kを破壊した(p85α-/-)マウスあるいは対照(p85α+/-)マウスから取り出し、Th17細胞に分化させた。Th17細胞の分化を示すIL-17A産生量は、p85α-/-細胞では対照(p85α+/-)に比べて低く抑えられている。
-
B:未分化なT細胞を野生型マウスから取り出し、クラスⅠA PI3K阻害剤であるIC87114処理(PBSとあるのは阻害剤を入れていないことを示す)を加えて、Th17細胞に分化させた。IL-17A産生量は、阻害剤処理によって低く抑えられている。
-
C:未分化なT細胞を野生型マウスから取り出し、mTORC1阻害剤であるラパマイシン処理(PBSとあるのは阻害剤を入れていないことを示す)を加えて、Th1あるいはTh17細胞に分化させた。Th1細胞の分化を示すIFNγ産生量に違いはないが、Th17細胞の分化を示すIL-17A産生量は、阻害剤処理によって低く抑えられている。
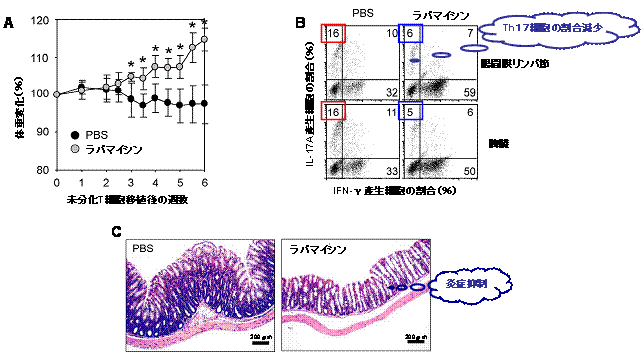
図2 ラパマイシンの投与によって大腸炎が軽減する
-
A:未分化なT細胞をT、B細胞欠損(Rag2-/-)マウスに移植すると、自己免疫性の大腸炎を発症して体重減少が観察されるが、ラパマイシンを連日投与することによって体重減少が見られなくなる。PBSとあるのは阻害剤を入れていないことを示す。
-
B:未分化なT細胞移植6週間後に、マウスの腸間膜リンパ節あるいは脾臓(ひぞう)に存在するTh1(IFNγ産生)細胞およびTh17(IL-17A産生)細胞の割合を調べた。対照(赤四角)(PBSとあるのは阻害剤を入れていないことを示す)に比べて、ラパマイシンの投与(青四角)によってTh17細胞の割合が減少した。
-
C:未分化なT細胞移植6週間後の大腸の組織像。対照(PBSとあるのは阻害剤を入れていないことを示す)では腸炎を発症して大腸の肥厚が見られるが、ラパマイシンの投与によって炎症が抑えられ、肥厚も見られない。
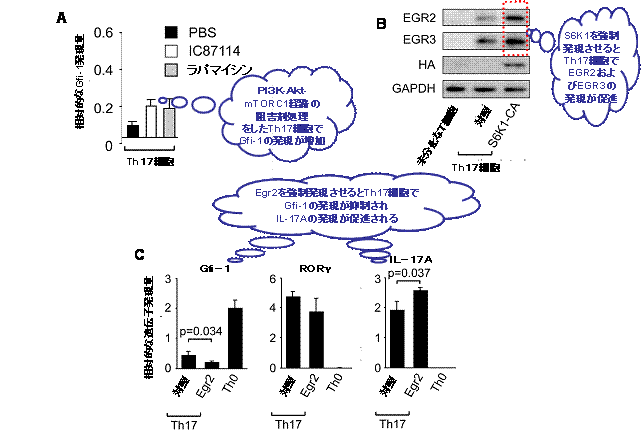
図3 PI3K-Akt-mTORC1経路の活性化は
Gfi-1の抑制を介してTh17細胞の分化を促進する
-
A:Th17細胞におけるGfi-1転写因子の発現はPI3K-Akt-mTORC1経路の阻害剤(IC87114あるいはラパマイシン)処理によって増強される。
-
B:mTORC1の下流で働くことが知られるS6K1転写因子の強制発現(S6K1-CA)により、未分化なT細胞をTh17細胞に分化させると、対照に比べてEGR2およびEGR3分子の発現が増強される。
-
C:未分化なT細胞でEGR2遺伝子を強制発現させることでTh17細胞に分化させ、Gfi-1、RORγ、IL-17Aの各遺伝子の発現量を調べた。EGR2遺伝子を強制発現させた細胞(Egr2)では、RORγ遺伝子の発現量には影響がないものの、対照に比べてGfi-1遺伝子の発現が抑制されるとともに、IL-17A遺伝子の発現が増強され、結果としてTh17細胞の分化が促進された。
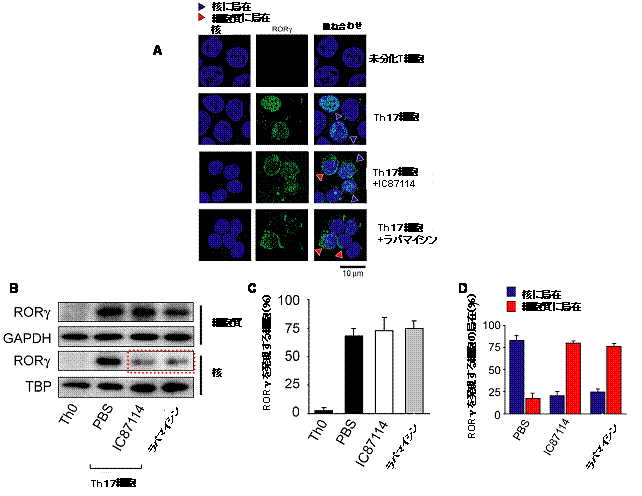
図4 Th17細胞の分化においてPI3K-Akt-mTORC1経路を阻害すると
RORγの核移行が阻害される
-
A:蛍光抗体法を用いたRORγ分子の細胞内分布。RORγはTh17細胞を特異的に発現して24時間以内に核に移行するが(青三角)、IC87114あるいはラパマイシンなどの阻害剤を加えておくと、RORγが細胞質にとどまっている様子が見られるようになる(赤三角)。
-
B:ウェスタンブロット法による細胞質内および核内に存在するRORγたんぱく質の検出。Th17細胞に発現するRORγは細胞質および核のどちらでも検出されるが、IC87114あるいはラパマイシンなどの阻害剤を加えると核で検出されるRORγたんぱく質の量が減少する。
-
C:RORγを発現する細胞の割合は、IC87114あるいはラパマイシンなどの阻害剤を加えても変わらない。
-
D:IC87114あるいはラパマイシンなどの阻害剤を加えることにより、核内に存在するRORγの割合(青)が減少し、その代わりに細胞質にとどまっているRORγの割合(赤)が増加する。
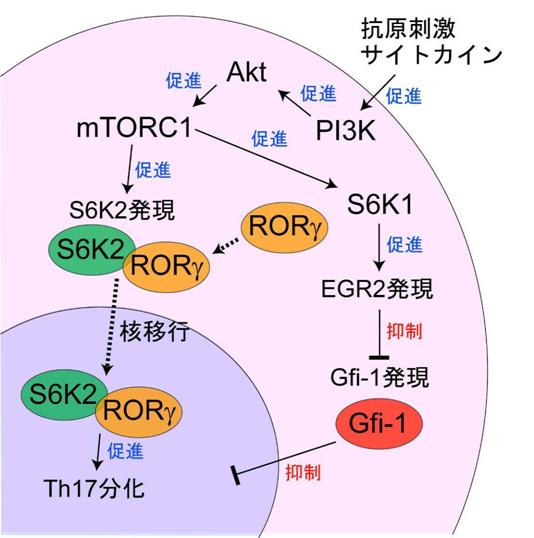
図5 研究成果のまとめ
未分化のT細胞が、抗原刺激とTh17細胞分化に必要なサイトカインの刺激を受け取ると、PI3K-Akt-mTORC1経路が活性化される。活性化されたmTORC1はS6K1とS6K2の発現を介して2通りの経路でTh17細胞の分化を促進する。すなわち、S6K1の発現を介してEGR2転写因子の発現を上昇させ、Th17細胞分化を阻害するGfi-1転写因子の発現を抑えることによって、結果的にTh17細胞の分化を誘導する一方で、S6K2は核移行シグナルとしてTh17細胞の分化に必須のRORγと結合し、核内に移行してTh17細胞の分化を促進する。
<用語解説>
- 注1) 関節リウマチ、炎症性腸疾患
- 関節リウマチは、免疫の異常によって関節に炎症がおこり、関節が腫(は)れて痛む病気。4倍程度女性に多く、発症が最も多い年齢は30~50歳代で、日本には約70万~100万人の患者がいると考えられている。
炎症性腸疾患は若年者に多く発症し、発症の原因不明、腸に炎症を起こす疾患。大きく2つに分けられ、1つは潰瘍性大腸炎(症状として下痢、下血、血便、粘液便などの排便異常が主体)、もう1つはクローン病(症状として、下痢・腹痛といった腹部の消化器の症状が中心)で、それぞれ約10万人、約3万人の患者がいると考えられている。炎症性腸疾患は西洋化した食事が一因といわれ、近年では日本における患者数が著しく増加している。
- 注2) Th17細胞
- 主にIL-17AやIL-17Fを産生することから名付けられた、ヘルパーT細胞の一種。細胞外寄生細菌などの排除に働く一方、自己免疫疾患の発症にも関与するとされている。転写因子RORγ(t)がこの細胞の分化に必須である。
- 注3) PI3K(phosphoinositide 3-kinase)
- 細胞膜の細胞質側に存在するホスファチジルイノシトールのイノシトール環の第3位をリン酸化する酵素。このPI3Kによって産生されるホスファチジルイノシトール3リン酸には、Aktをはじめ、さまざまな分子が結合して細胞膜面に集まり、シグナル反応の場を形成することによって、細胞分裂や代謝などさまざまな細胞機能を調節する。
- 注4) サイトカイン
- 免疫細胞をはじめとする細胞から分泌され、または作用するたんぱく質で、免疫・造血・炎症反応などの制御に関与する情報伝達を司る微量たんぱく質の総称。インターロイキンやインターフェロン、TGFβなども含む。
- 注5) 転写因子
- DNAに結合することでそのDNAの発現を促進または抑制して調節するたんぱく質のこと。ヒトでは約1800種類あると考えられている。
- 注6) たんぱく質リン酸化酵素
- たんぱく質をリン酸化させる酵素のことで、ヒトでは約500種類あると考えられている。たんぱく質はこのリン酸化によって酵素活性やほかのたんぱく質との会合状態を変化させ、さまざまなシグナル伝達を行っている。
- 注7) mTORC1(mTOR complex 1)
- セリン・スレオニンキナーゼ(たんぱく質のセリン残基あるいはスレオニン残基にリン酸基を付加する酵素)の1つであるmTOR(mammarian target of rapamycin)と呼ばれるたんぱく質が、Raptor、GβL(mLST8)、PRAS40、Deptorと会合してできた複合体。増殖、栄養、あるいはエネルギー代謝など多くのシグナルを統合している。PI3K-Akt経路からは、TSC1/2、Rhebを介してmTORC1へとシグナルが伝わる。mTORC1が活性化されると、下流に存在するp70S6K(S6K)や4E-BP1をリン酸化し、その結果として、リボゾームたんぱくS6(たんぱく合成)やeIF4E(転写調節)の機能を促進させる。
- 注8) ラパマイシン(rapamycin)
- イースター島の土壌サンプルから発見された放線菌「Streptomyces hygroscopics 」から産生される抗生物質で、免疫抑制剤の1つ。シロリムス(Sirolimus)とも呼ばれる。mTORC1の酵素活性を選択的に阻害する。
<論文名>
“PI3K-Akt-mTORC1-S6K1/2 axis controls Th17 differentiation by regulating Gfi-1 expression and nuclear translocation of RORγ.”
(PI3K-Akt-mTORC1-S6K1/2経路はGfi-1の発現およびRORγの核内移行を調節することによってTh17細胞分化を制御する)
doi: 10.1016/j.celrep.2012.02.007
<お問い合わせ先>
<研究に関すること>
永井 重徳(ナガイ シゲノリ)
慶應義塾大学 医学部 微生物学・免疫学教室 助教
〒160-8582 東京都新宿区信濃町35 東校舎3階
Tel:03-5363-3769 Fax:03-5361-7658
E-mail:
<JSTの事業に関すること>
石井 哲也(イシイ テツヤ)
科学技術振興機構 イノベーション推進本部 研究領域総合運営部
〒102-0076 東京都千代田区五番町7 K’s五番町
Tel:03-3512-3524 Fax:03-3222-2064
E-mail: