JST 課題解決型基礎研究の一環として、慶應義塾大学 医学部の吉村 昭彦 教授らは、自己免疫疾患の発症を抑える新たな免疫調節メカニズムを発見しました。関節リウマチや多発性硬化症注1)などの自己免疫疾患の発症は、免疫系の司令塔の役割をするT細胞のなかでも、最近発見されたTh17細胞注2)が中心的な役割を果たしていると考えられています。そのため、自己免疫疾患の治療標的としてTh17細胞をめぐる研究が世界中で盛んに行われていますが、Th17細胞がどのようなメカニズムで産み出されるかは不明でした。
本研究グループは今回、Th17細胞の誘導を制御する重要な経路、すなわち未熟なT細胞からTh17細胞になるためにはEomesodermin(Eomes:エオメス)注3)と呼ばれるたんぱく質が減少しなければならないことを実験で明らかにしました。他の種類のT細胞ではEomesの発現は高く維持されており、Eomesを未熟なT細胞に人為的に発現させるとTh17細胞になりにくいことを確認しました。また、Eomesを減少させるために必要な酵素がJNK注4)と呼ばれるリン酸化酵素であることも突き止めました。JNKの阻害剤を脳脊髄系の自己免疫疾患である多発性硬化症のモデルマウスに投与したところ、症状が著しく改善しました。
これらの発見をもとに、T細胞でEomesの発現を高めるか、あるいはJNKの活性を抑制することで、リウマチや多発性硬化症などの自己免疫疾患に対する新しい治療方法の開発につながるものと期待されます。
本研究成果は、2011年5月19日(米国東部時間)に米国科学雑誌「Immunity」のオンライン速報版で公開されます。
本成果は、以下の事業・研究領域・研究課題によって得られました。
戦略的創造研究推進事業 チーム型研究(CREST)
| 研究領域 |
「アレルギー疾患・自己免疫疾患などの発症機構と治療技術」
(研究総括:菅村 和夫 宮城県立病院機構 理事長) |
| 研究課題名 |
「細胞内シグナル制御による免疫リプログラミング」 |
| 研究代表者 |
吉村 昭彦(慶應義塾大学 医学部 微生物学・免疫学教室 教授) |
| 研究期間 |
平成20年10月~平成26年3月 |
JSTはこの領域で、アレルギー疾患や自己免疫疾患を中心とするヒトの免疫疾患を予防・診断・治療することを目的に、免疫システムを適正に機能させる基盤技術の構築を目指しています。
上記研究課題では、細胞内のシグナル伝達制御機構の解明とその人為的な調節により新たな免疫疾患治療の方法論を開発することを目指しています。
<研究の背景と経緯>
免疫システムは私たちの身体を多種多様な病原体から守っています。ヘルパーT(Th)細胞は免疫系の司令塔としての役割を担っており、侵入した病原体の種類に応じて、Th1、Th2、Th17の3種類のいずれかのT細胞に分化誘導され、その病原体の排除に最適な免疫応答を誘導します(図1)。一方でこれらが過剰に作用したり、自己のたんぱく質などに反応すると免疫疾患になります。例えば、Th2細胞が過剰に産生されると、花粉や食物などに反応してアレルギー疾患を引き起こします。またTh17細胞は最近発見されたT細胞ですが、関節炎リウマチや多発性硬化症、乾癬、炎症性腸疾患などの自己免疫疾患を引き起こす細胞として注目されています(参考文献1、2)。そして、Th17細胞を抑制することで多くの自己免疫疾患を治療できるのではないかと期待されています。
未熟なT細胞がTh17細胞へと成熟分化するにはインターロイキン注5)6(IL‐6)や腫瘍増殖因子β(TGFβ)と呼ばれるサイトカイン注6)が必要であることが知られていました(図1)。TGFβはRORγtと呼ばれる転写因子の発現促進を介して、Th17細胞への分化を誘導することは分かっていましたが、その詳細な仕組みは不明でした。もしその仕組みが分かれば、Th17細胞を抑制する有効な方法が発見できるであろうと考えられてきました。そこで本研究グループではTh17細胞が産み出されるメカニズムを解明しようと試みました。
<研究の内容>
本研究グループはまず、TGFβによって発現に影響が出る遺伝子のなかで、Th17細胞への分化と関連する遺伝子を検索しました。影響が出た遺伝子のなかで、Eomesと呼ばれるたんぱく質を作る遺伝子がTh17細胞では発現を抑えられ、他のTh1細胞やTh2細胞では抑えられないことを突き止めました(図2A)。そこで、Eomesを未熟なT細胞に人為的に発現させたところ、Th17細胞の誘導が著しく減少していました(図2B)。逆にEomesの発現を抑えたT細胞ではTGFβを加えなくてもTh17細胞が誘導されました(図2C)。Eomesは転写因子としてDNAに結合して遺伝子の発現を制御するたんぱく質です。さまざまな実験からEomesは、Th17分化に最も重要なRORγtやIL‐17Aの遺伝子のプロモーターと呼ばれる部分に作用して発現を抑制していることが分かりました(図3)。
次に、T細胞でどのようなシグナルがEomesの発現をコントロールしているのかを調べました。TGFβにはいくつかのシグナル伝達経路が知られていますが、そのなかでJNKと呼ばれるリン酸化酵素はTGFβによって活性化されることが知られていました。このJNKの阻害剤を加えてJNKを働かなくさせると、TGFβによるEomesの発現抑制が解除されることが分かりました(図4A)。またJNKを阻害することでIL‐17Aの発現、すなわちTh17細胞分化が抑制されること(図4B)や、JNKの発現を低下させるとEomesの発現が高くなりTh17細胞が減少することも分かりました。逆にJNKをT細胞に過剰に発現させるとEomesの発現量が少なくなり、TGFβがなくてもTh17細胞分化が誘導されていました。
次にJNK阻害剤を多発性硬化症のモデルマウスである実験的自己免疫性脳脊髄炎モデル注7)に適用してみました。JNK阻害剤は予想通りTh17細胞分化を抑制し、実験的自己免疫性脳脊髄炎の症状を著しく改善しました(図5)。
JNKは、c‐Junと呼ばれる転写因子を介して機能を発揮することが知られています。詳細な検討から、やはりc‐JunがEomesたんぱく質を作る遺伝子の発現を低下させることが分かりました。すなわち、TGFβ → JNK → c‐Jun → Eomesの抑制 → Th17細胞分化誘導というTh17細胞分化を制御するメカニズムがあることが明らかになりました(図6)。
本研究グループは今回の結果から、Th17細胞分化を調節する新たなメカニズムを明らかにし、自己免疫疾患治療の標的となる酵素を発見しました。
<今後の展開>
本研究によってJNK‐Eomes経路はTh17細胞分化を誘導することが明らかになりました。よってEomesたんぱく質の発現やJNKの機能調節により、さまざまな免疫疾患の制御が可能になると考えられます(図6)。しかし、TGFβがTh17細胞を誘導するメカニズムはまだ全容が解明されたわけではありません。今後も引き続きTGFβがTh17細胞分化を誘導する機構の全容を明らかにするとともに、TGFβのシグナルを標的とした自己免疫疾患の治療法の開発を進めたいと考えています。
<参考図>
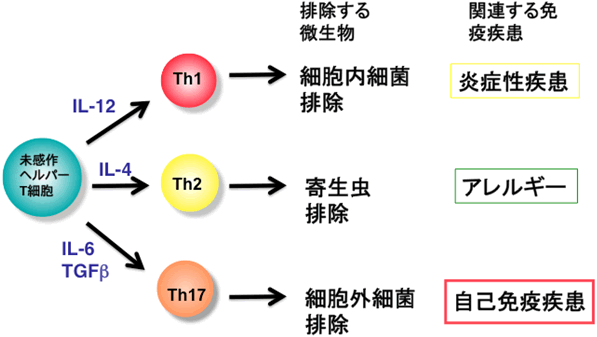
図1 ヘルパーT細胞の分化と関連する免疫応答と免疫疾患
侵入した病原体の種類に応じて、3種類いずれかのエフェクターヘルパーT細胞(Th1、Th2、Th17)が未感作ヘルパーT細胞から分化し、その病原体の排除に最適な免疫応答を誘導する。Th1細胞誘導にはインターロイキン-12(IL‐12)、Th2細胞誘導にはIL‐4、Th17細胞誘導にはTGFβとIL‐6といったサイトカインが分化の方向付けを行う。一方、これらの細胞が過剰に応答したり、自己たんぱく質などに反応するとさまざまな疾患が発症する。このなかでTh17細胞は自己免疫疾患と特に関係が深いと考えられている。
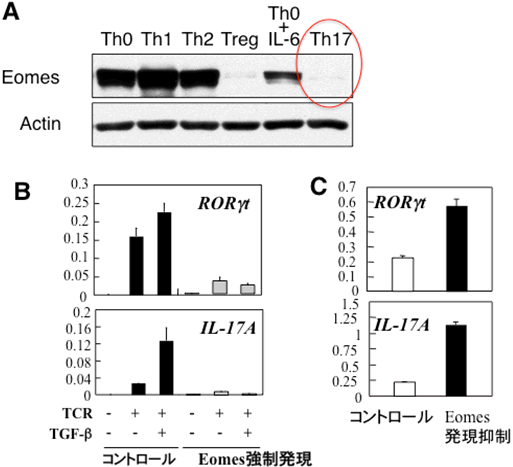
図2 Eomesの発現量が少ないと、Th17細胞分化が誘導される
- A: 未熟なT細胞をそれぞれのヘルパーT細胞に分化させて、Eomesの発現を検出した。Th0は中性条件でいずれのヘルパーT細胞にもならない条件。制御性T細胞(Treg)はTGFβとIL‐2で、Th17細胞はTGFβとIL‐6で誘導した。赤で囲ってあるTh17やTregは誘導にTGFβが使われているが、いずれもEomesの発現が低く抑えられている。
- B: ヘルパーT細胞にEomesを強制的に発現させた場合のTh17細胞分化。T細胞にEomesを強く発現させた場合、T細胞の刺激(TCR)やTGFβ刺激によって誘導され、Th17細胞に特徴的な遺伝子でもあるRORγtやIL‐17Aの発現が低く抑えられ、Th17細胞分化が抑制される。
- C: ヘルパーT細胞でEomesを人為的に発現させなくした場合のTh17細胞分化。Eomesの発現を抑制するとRORγtやIL‐17Aの発現が亢進することが分かる。この時、TGFβを加えていないのでEomesの抑制はTGFβを代替できることも分かる。
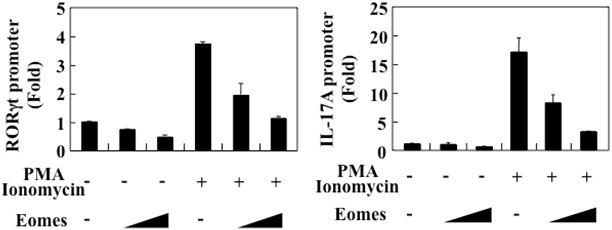
図3 EomesはRORγtとIL‐17Aの転写を抑制する
RORγt(左)とIL‐17A(右)の遺伝子の発現をコントロールするプロモーター(promoter)と呼ばれる部分の転写活性を調べた。これらのプロモーターは、T細胞の刺激を模倣するPMAとIonomycinという薬剤で活性化されるが、その時にEomesを発現させるとプロモーターの活性が抑制されることが分かる。このことから、EomesはRORγtとIL‐17Aのプロモーターに作用してTh17細胞分化を抑制することが明らかになった。
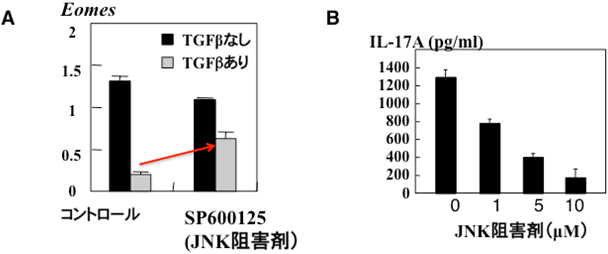
図4 JNK阻害剤はEomesの発現を回復させTh17細胞の分化を抑制する
- A: Th17細胞分化誘導時にJNK阻害剤SP600125を添加すると、TGFβによるEomesの発現抑制が解除される。
- B: 同時にJNK阻害剤はIL‐17Aの発現(Th17細胞分化)を抑制する。
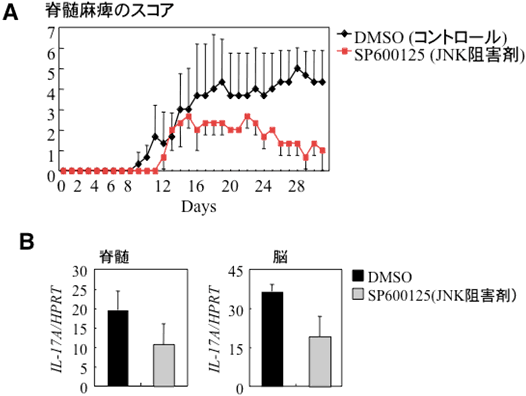
図5 JNK阻害剤は実験的自己免疫性脳脊髄炎を抑制する
- A: マウスを用いた実験的自己免疫性脳脊髄炎のモデル。JNK阻害剤は免疫開始後2日後より11日目まで毎日0.5mg/kgを腹腔内に投与した。脊髄麻痺の状況をスコア化している。
- B: 実験的自己免疫性脳脊髄炎誘導後31日目の脊髄と脳でのIL‐17Aの発現。JNK阻害剤で抑制されていることが分かる。
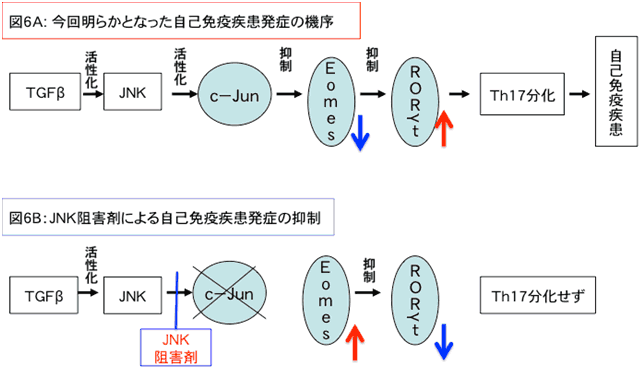
図6 JNK‐Eomes経路による自己免疫疾患発症のメカニズムとJNK阻害による治療原理
- A: JNKがTGFβによって活性化し、c-Junを活性化するとEomesの発現が抑制される。結果としてRORγtが発現し、Th17細胞分化が誘導されて自己免疫疾患を発症する。
- B: JNK阻害剤によってJNKの働きが阻害されると、c-Junの活性化が抑制され、Eomesの発現が上昇する。その結果EomesはRORγtの発現を抑制し、Th17細胞分化が誘導されず、自己免疫疾患の発症を抑える。従って、JNK阻害剤もしくはEomesの発現誘導は自己免疫疾患の治療に結びつくと考えられる。
<用語解説>
- 注1) 多発性硬化症
- 中枢性脱髄疾患の1つで、脳、脊髄、視神経などに病変が起こり、多彩な神経症状が再発と寛解を繰り返す。自己反応T細胞や抗体により神経の髄鞘が破壊されることで神経麻痺などが起こると考えられている。日本では特定疾患に認定されている指定難病である。
- 注2) Th17細胞
- ヘルパーT細胞の一種で、IL‐17AやIL‐17F、IL‐22といったサイトカインを分泌し、主に細胞外の細菌や真菌の排除に働く。一方で自己免疫疾患にも深く関与することが知られている。Th17細胞を作る最も重要な転写因子がRORγtである。
- 注3) Eomesodermin(Eomes)
- T‐box転写因子族の一種で、脊椎動物の発生や分化にかかわる。T細胞においてはこれまでNK細胞やCD8キラーT細胞での機能が知られていたが、ヘルパーT細胞においては今回Th17細胞分化の抑制に働くことが明らかになった。
- 注4) JNK(c-Jun N-terminal kinase)
- たんぱく質リン酸化酵素の一種で、細胞内外のさまざまな刺激で活性化され、c‐Junと呼ばれる転写因子をリン酸化する。リン酸化されたc‐Junはc‐Fosなど他の転写因子と複合体を作ってDNAに結合してさまざまな遺伝子の転写を制御する。
- 注5) インターロイキン
- 細胞から分泌されるたんぱく質であるサイトカインの一種で、主にマクロファージやリンパ球から産生される。インターロイキンはリンパ球間の情報を伝達する重要な因子で免疫系では感染防御のほか、さまざまな免疫疾患に関与する。
- 注6) サイトカイン
- 免疫細胞から分泌されるたんぱく質で、特定の細胞に免疫や炎症をはじめ、分化や細胞死などの情報を伝達する役割を持つ。インターロイキンの他、インターフェロンやTGFβもサイトカインに含まれる。例えば、未熟なT細胞はTGFβとIL-6で刺激されることによりTh17細胞に分化する。
- 注7) 実験的自己免疫性脳脊髄炎モデル
- 多発性硬化症の動物モデルとして知られている。マウスやラットを髄鞘構成糖たんぱく質(MOG)と免疫機能を高めるアジュバント(免疫増強剤)を注射して免疫することにより、多発性硬化症に似た脳脊髄炎を発症する実験モデル。
<論文名>
“Transcription factor Smad-independent T helper 17 cell induction by Transforming-Growth factor-b is mediated by suppression of Eomesodermin”
(転写因子Smad非依存性17型ヘルパーT細胞の誘導はEomesoderminの抑制によって仲介される)
<参考文献>
参考文献1:
Cua DJ et al.
“Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain”
Nature. 421, 744-748 (2003)
参考文献2:
Park H et al.
“A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17”
Nat Immunol. 6, 1133-1141 (2005)
<お問い合わせ先>
<研究に関すること>
吉村 昭彦(ヨシムラ アキヒコ)
慶應義塾大学 医学部 微生物学・免疫学教室 教授
〒160-8582 東京都新宿区信濃町35 東校舎4F
Tel:03-5363-3483 Fax:03-5360-1508
E-mail:
<JSTの事業に関すること>
石井 哲也(イシイ テツヤ)
科学技術振興機構 イノベーション推進本部 研究領域総合運営部
〒102-0075 東京都千代田区三番町5 三番町ビル
Tel:03-3512-3524 Fax:03-3222-2064
E-mail: